1.1?植物組織:CTAB法
- 液氮研磨樣品,分裝至離心管中
- 向離心管中加入 CTAB,水浴
- 離心后向上清中加入氯仿/異戊醇(24:1),離心
- 向上清中加入異丙醇和乙酸鈉溶液,離心,棄上清
- 加入 75%乙醇,離心,棄上清
- 晾干,加入 TE ?溶解 DNA ,保存在-20℃冰箱
1.2?動物組織:SDS法
- 液氮研磨組織樣,分裝至離心管中
- 加入 SDS 溶液,水浴
- 加入 NaCl 溶液,靜置后離心
- 向上清中加入氯仿/異戊醇(24:1)后離心
- 向上清中加入異丙醇后離心,棄上清
- 加入 75%乙醇,離心,棄上清
- 晾干,加入 TE 溶解 DNA,保存在-20℃冰箱
1.3?動物血液:Kurabo 試劑盒法
QuickGene DNA whole blood kit S (DB-S),廠家:Kurabo,貨號:40321300101
1.4?粗體核酸純化
試劑盒名稱:QIAGEN Genomic-tip 20/G,廠家:QIAGEN,貨號:10223
試劑盒名稱:QIAGEN Genomic-tip 100/G,廠家:QIAGEN,貨號:10243
1.5?微生物樣本
使用TGuide S96磁珠法土壤/糞便基因組DNA提取試劑盒完成核酸的提取
2、檢測方法
2.1?檢測方法
- 濃度檢測
Nanodrop:賽默飛 Thermo ,型號 NANODROP2000
Qubit:廠家YEASEN, 型號CAT 12642-A ,試劑1XdsDNA HS Assay kit for Qubit
- 完整性檢測(瓊脂糖凝膠電泳)
電泳儀:廠家:天能 Tanon ,型號 EPS600
電泳槽:廠家:天根生化科技(北京)有限公司,型號:HE- 120
2.2?判定標準
- 總量≥2μg(單次建庫)
- 濃度≥50ng/ul
- 體積≥10(ul)
- OD260/280在7-2.2之間,OD260/230在1.0-3.0之間
- AGE:size≥23K,降解條帶>5K,點樣孔無或有輕微污染,N/Q在8-2.5之間
- 樣本狀態正常,樣本粘稠、顏色異常、有渾濁或不溶物均不合格
注:特殊物種實驗人員會根據經驗進行判斷,具體以檢測報告中的判斷結果為準
3、建庫方法
3.1?建庫流程
- 準備高質量核酸,g-TUBE 管使核酸片段化
- 使用BluePippin進行片段篩選
- 使用 NEBNext FFPE DNA Repair Mix 和 NEBNext Ultra II End Repair/dA-Tailing Module 完成核酸片段的損傷修復和末端修復加 A
- 使用無擴增條形碼擴展試劑盒添加 barcode 序列
- 使用無擴增條形碼擴展試劑盒完成測序接頭的連接
3.2?建庫試劑
- 文庫構建試劑:NEBNext FFPE DNA Repair Mix貨號?M6630L 廠家?NEB
- 文庫構建試劑:NEBNext Ultra II End Repair/dA-Tailing Module貨號E7546L ?廠家NEB
- 文庫構建試劑:無擴增條形碼擴展試劑盒1-96 ?貨號SQK-NBD114.96 廠家Nanopore
3.3?建庫設備
- 數字 3*32PCR 儀??廠家?Appliedbiosystems???儀器規格?proflex3*32
- 制冷型程控五段金屬浴??廠家?天根生化科技(北京)有限公司??儀器規格?OSE-DB-02
4、測序方法
4.1?上機操作
- 用測序芯片制備試劑盒配制 Flow cell Priming mix
- 用測序輔助擴展試劑盒配置上機文庫
- 用 PromethION ?Flow ?Cells 芯片,在PromethION48測序儀運行MinKnow 軟件,開始測序,默認運行72 小時
4.2?測序試劑
- 測序芯片制備試劑盒??貨號?EXP-FLP004 ??廠家?Nanopore
- 測序輔助擴展試劑盒??貨號?EXP-AUX003 ?廠家?Naonopore
- 測序芯片??PromethION Flow Cell(R10.4.1)??貨號?FLO-PRO114M ??廠家?Nanopore
4.3?測序設備
PromethION48 測序儀??廠家?Nanopore???儀器規格?PromethION48
]]>發表期刊:中國農業科學
發表時間:2020年11月
影響因子:2.302
研究背景
蜜蜂球囊菌(Ascosphaeraapis,簡稱球囊菌)是專性侵染蜜蜂幼蟲的致死性真菌病原,引發的白堊病是長期危害養蜂生產的頑疾,不僅可導致蜜蜂幼蟲的大量死亡,還能導致成年蜜蜂數量的銳減以及蜂群群勢和蜂產品產量的驟降。目前,球囊菌的基因組注釋信息尚不完善,高質量參考轉錄組匱乏,嚴重限制了球囊菌的組學和分子生物學研究。
材料和方法
球囊菌菌株由福建農林大學動物科學學院(蜂學學院)蜜蜂保護實驗室分離、純化和保存。純化得到的純凈菌絲樣品和孢子樣品經液氮速凍后迅速轉移到-80℃超低溫冰箱保存備用。利用納米孔長讀段測序技術對球囊菌的純化菌絲(Aam)和純化孢子(Aas)分別進行測序,將高質量的三代測序數據混合后用于構建全長轉錄組,并通過比對主流數據庫進行功能注釋,同時對球囊菌的長鏈非編碼RNA(longnon-codingRNA,lncRNA)進行鑒定和分析。
結果
1、納米孔測序數據質控
球囊菌菌絲和孢子的納米孔測序分別得到6321704和6259727條原始讀段,N50分別達到1094和1157bp,平均長度分別為992和1047bp,長的長度分別為9421和13060bp(表1)。來源于Aam和Aas的原始讀段的長度分布介于1-10kb以上,其中分布reads數多的長度均為1kb(圖1-A、1-B);原始讀段的Q值分布介于Q6-Q15,分布reads數多的質量值分別為Q9和Q11(圖1-C、1-D)。
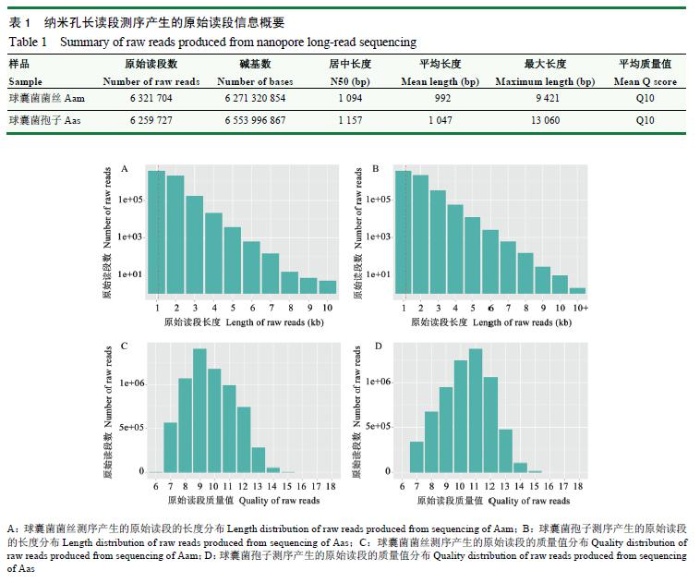
圖1球囊菌菌絲和孢子納米孔長讀段測序的原始讀段長度和質量值分布Fig.1Lengthandqualitydistributionofrawreadsgeneratedfromnanoporelong-readsequencingofA.apismyceliumandspore
2、全長轉錄本的鑒定和分析
進一步過濾冗余全長有效讀段,分別得到9859和16795條非冗余全長轉錄本,N50分別達到1482和1658bp,平均長度分別達到1187和1303bp,長的長度分別為6472和6815bp(表2);上述非冗余全長轉錄本的長度介于1-7kb,其中分布在1kb的全長轉錄本數多。進一步對Aam和Aas的非冗余全長轉錄本進行Venn分析,結果顯示有6512個非冗余全長轉錄本為菌絲和孢子所共有,分別有3347和10283個非冗余全長轉錄本為二者特有(圖2-A)。
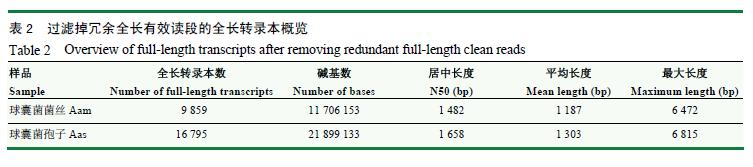
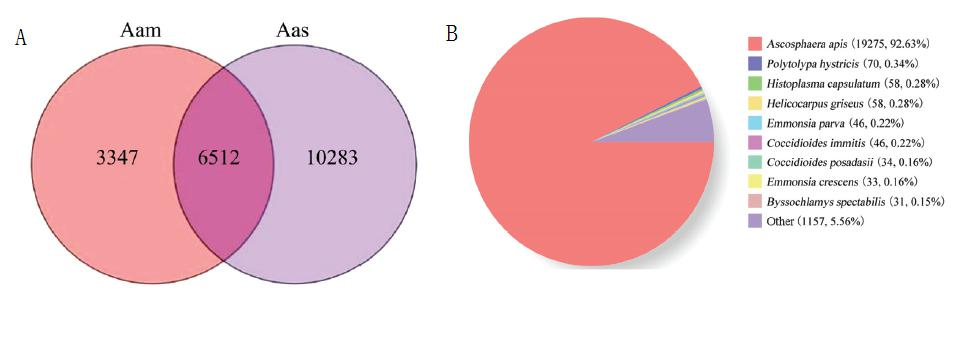
圖2球囊菌菌絲和孢子全長轉錄本的Venn分析(A)、全長轉錄本的Nr數據庫注釋(B)Fig.2Vennanalysisoffull-lengthtranscriptsinA.apismyceliumandspore(A)、Nrdatabaseannotationoffull-lengthtranscripts(B)
3、全長轉錄本的數據庫注釋
在球囊菌菌絲和孢子中共鑒定出20142條全長轉錄本,數據庫注釋結果顯示,分別有20809、11151、17723、12164、11340和9833全長轉錄本可注釋到Nr、KOG、eggNOG、Pfam、GO和KEGG數據庫。注釋全長轉錄本數量多的物種是球囊菌、Polytolypahystricis和莢膜組織胞漿菌(Histoplasmacapsulatum)(圖2-B)
4、lncRNA的鑒定及分析
利用CPC、CPAT、CNCI和Pfam4種方法依次鑒定出1906、1682、750和648條lncRNA,四者的交集為648個(圖3-A);其中基因間區lncRNA(longintergenicRNA,lincRNA)、反義鏈lncRNA(anti-senselncRNA)和正義鏈lncRNA(senselncRNA)的數量分別為480、119和49個(圖3-B)。
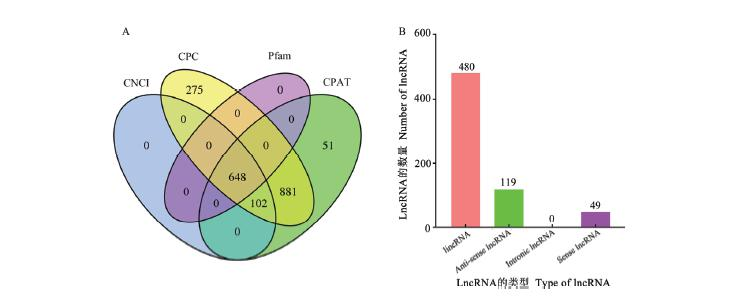
圖 3 球囊菌 lncRNA 的數量(A)和種類(B) Fig. 3 Number (A) and type (B) of A. apis lncRNAs
總結
構建和注釋了球囊菌的高質量全長轉錄組,為探究球囊菌轉錄組的復雜性、完善參考基因組的序列和功能注釋信息以及深入開展球囊菌可變剪接體的功能研究提供了關鍵依據。
深度挖掘數據和拓展
同期作者利用納米孔全長轉錄組測序數據對蜜蜂球囊菌(Ascosphaeraapis)和另一蜜蜂真菌病原東方蜜蜂微孢子蟲(Nosemaceranae)的現有參考基因組在結構功能注釋上進行了較好的完善,同時也對基因的可變剪接(alternativesplicing,AS)和可變多聚腺苷酸化(alternativepolyadenylation,APA)進行解析。通過gffcompare軟件將全長轉錄本與參考基因組注釋的轉錄本進行比較,對基因組注釋基因的非編碼區向上游或下游延伸,修正基因的邊界。利用MISA軟件鑒定長度在500bp以上的全長轉錄本的簡單重復序列(simplesequencerepeat,SSR)位點信息。使用Blast工具將鑒定到的新基因和新轉錄本比對Nr、KOG、eggNOG、GO和KEGG數據庫,從而獲得功能注釋。通過Astalavista軟件鑒定基因的AS事件類型,統計分析可變剪切的結果。采用TAPISpipeline對基因的APA位點進行鑒定,得到APA的位點信息。分別利用CPC、CNCI、CPAT、Pfam4種方法對長鏈非編碼RNA(longnon-codingRNA,lncRNA)進行預測,取四者的交集作為高可信度的lncRNA。研究結果較好地優化了現有的東方蜜蜂微孢子蟲和蜜蜂球囊菌參考基因組已注釋基因的結構和功能注釋信息,并補充和注釋了大量參考基因組未注釋的新基因和新轉錄本,同時也為其他真菌的AS和APA研究提供了有益的思路和方法借鑒。
]]>英文題目:Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain
發表雜志:Mol. Psychiatry,2020年1月
影響因子:11.973
研究背景
在人腦中,與精神分裂癥相關的基因組區域富集了在神經發育過程中表現出不同異構體使用的基因,RNA剪接是將遺傳變異與精神疾病聯系起來的關鍵機制。剪接圖譜在大腦中特別多樣,很難準確識別和量化。短讀長RNA-Seq方法不能準確地重建和定量大多數轉錄物和蛋白質異構體,為解決這一挑戰,本文將long-range PCR和nanopore全長轉錄組測序與一種新的生信分析流程結合。
CACNA1C是一種精神危險基因,編碼電壓門控鈣通道CaV1.2,CACNA1C基因很大而且很復雜,至少有50個注釋外顯子和31個預測的轉錄本。它的大小和復雜性使得用標準的基因表達方法準確鑒定和量化轉錄本變得極其困難,本文在人腦中鑒定了CACNA1C的全長編碼轉錄本,識別了38個新的外顯子和241個新的轉錄本,對異構體多樣性的詳細了解對于將精神病學基因組發現轉化為病理生理學見解和新的精神藥理靶點至關重要。
研究方法
樣本:來自利伯腦發育研究所儲存庫的三名成年捐贈者的尸檢腦組織(提取小腦、紋狀體、背外側前額葉皮質、扣帶回、枕葉和頂葉皮質的RNA,并進行逆轉錄)
測序方法:使用PCR擴增CACNA1C全長CDS,使用MinION進行測序
分析流程:https://github.com/twrze/TAQLoRe
研究結果
1、CACNA1C有很多外顯子和異構體
由于CACNA1C的復雜性,本文使用了兩種互補的方法來鑒定轉錄本:外顯子水平和剪接位點水平的分析,分析流程見補充圖2。該方法共鑒定了251種存在于人腦中獨特的CACNA1C轉錄異構體,其中241種是新的,包括使用新的外顯子,新的剪接位點和連接。
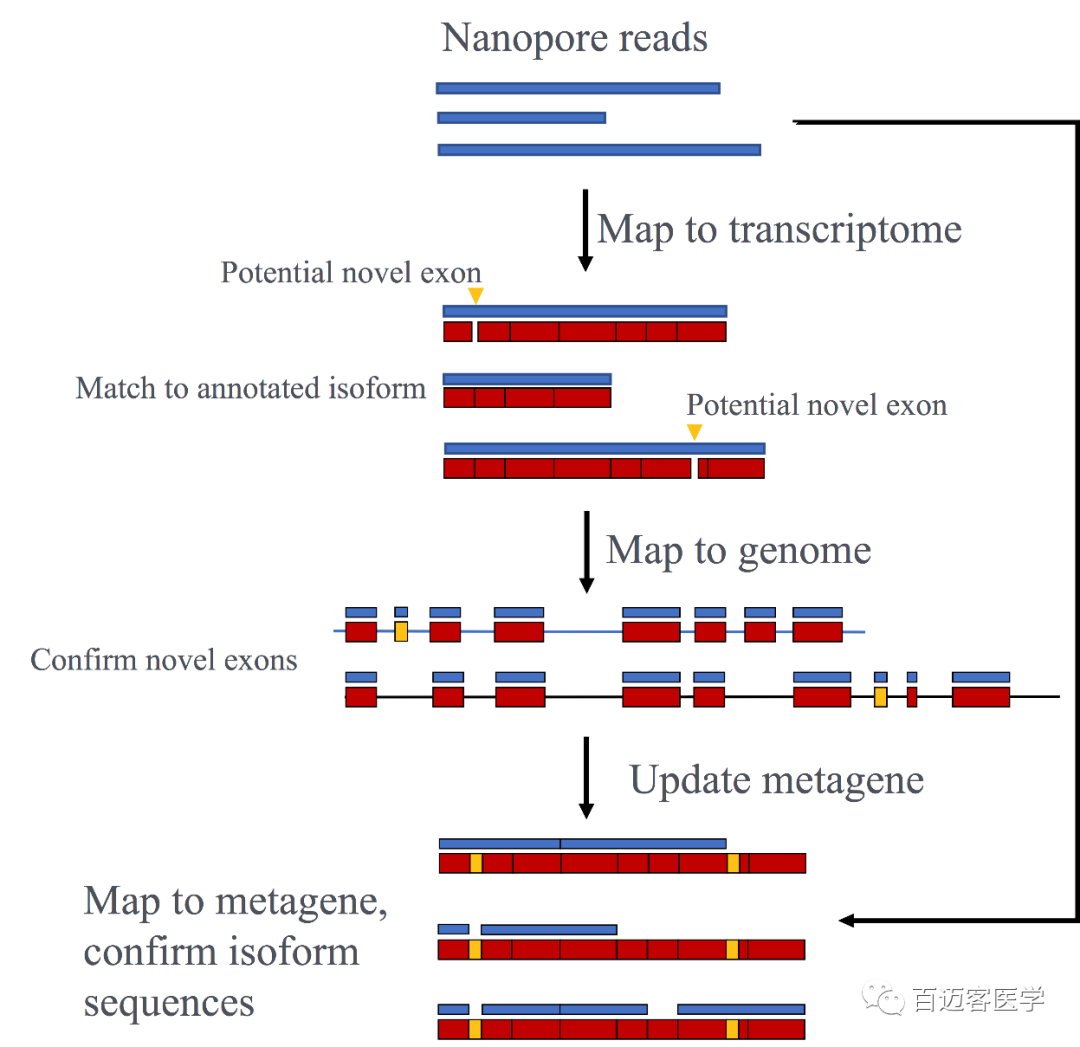
補充圖2
在CACNA1C基因座內總共注釋了39個潛在的新外顯子,其中38個在至少2個人或組織中被識別,并在每個文庫中得到至少5條nanopore reads的支持(圖2A)。通過PCR和Sanger測序確認了新的外顯子與其周圍的注釋外顯子之間的剪接連接,從而驗證了四個新的外顯子。這種新的外顯子的成功驗證提供了很高的可信度,即通過納米孔測序鑒定的新的外顯子是真實的,并且被整合到CACNA1C轉錄本中。表達量最高的10條轉錄本中,有9條是新的且其中有8條被預測保持CACNA1C閱讀框架,這表明這些最豐富的新轉錄本中有一些編碼功能不同的蛋白質異構體(圖2B,C)。這些結果表明,新的CACNA1C轉錄本表達豐富,數量也很多,目前的注釋缺少許多最豐富的CACNA1C轉錄本。
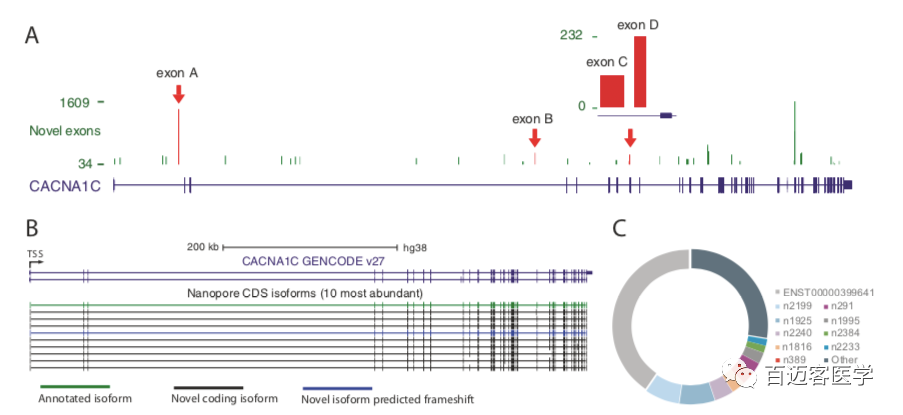
圖2
通過設置轉錄本的高置信度,在6個大腦區域確定了90個高可信的CACNA1C轉錄本,包括7個先前注釋的(GENCODE V27)和83個新的(補充圖3)。7個新的高置信度轉錄本包含新的外顯子,而其余76個包含以前未描述的連接和連接組合。
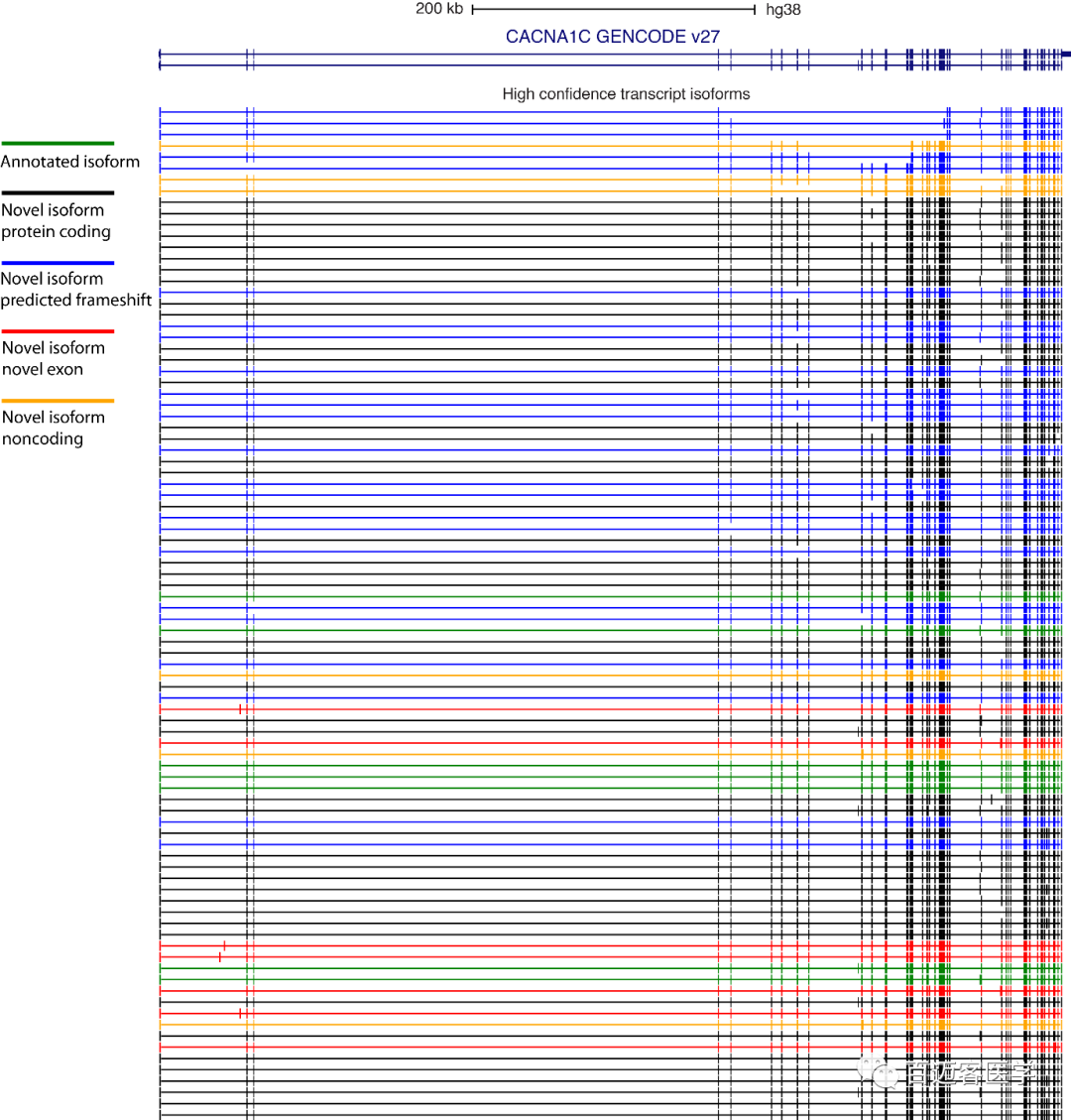
補充圖3
上述外顯子水平的轉錄本鑒定方法為鑒定新的外顯子和表征全長轉錄本結構提供了穩健和保守的手段。使用了更為保守的依賴于連接處無錯誤映射所支持的連接的識別,以及規范剪接位點的方法,確定了497個新的剪接位點,其中393個由至少10條reads支持,這些剪接位點,在篩選了至少24條reads支持的轉錄本后,鑒定了195個轉錄本,其中111個被預測為編碼的。
2、CACNA1C亞型在不同腦區的表達譜不同
小腦、紋狀體與皮質等組織觀察到了CACNA1C轉錄本差異,但在不同個體之間的表達是相似的。在小腦中觀察到了明顯的轉錄本表達轉換;在小腦之外,ENST00000399641是主要的轉錄本,而在小腦中,ENST00000399641和CACNA1C n2199的表達水平相似。
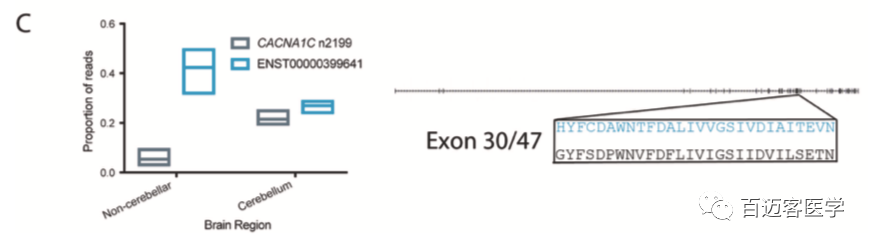
圖3 C
3、預測新isoforms對CaV1.2蛋白模型的影響
CACNA1C編碼CaV1.2 的主要成孔亞基。鈣孔由24個跨膜重復序列組成,由細胞內環連接成4個結構域(I-IV)(圖4A)。在我們鑒定的83個新的外顯子水平的轉錄本中,51個可能編碼功能性的CaV1.2通道。灰色方框表示新的、框架內的插入和刪除的位置(值表示包含每個isoforms的reads的平均比例)。使用兩種分析方法(外顯子水平和剪切連接水平)鑒定變體的情況,外顯子水平計數用于得出豐度(紅色文本);僅使用剪接位點水平方法鑒定的變體用藍色文本表示。包含三個微缺失的蛋白質異構體的數量:(I)在I-II接頭中,(Ii)在IV4-5接頭中,以及(Iii)在IV3-4接頭中先前報道的微缺失(圖4B)。
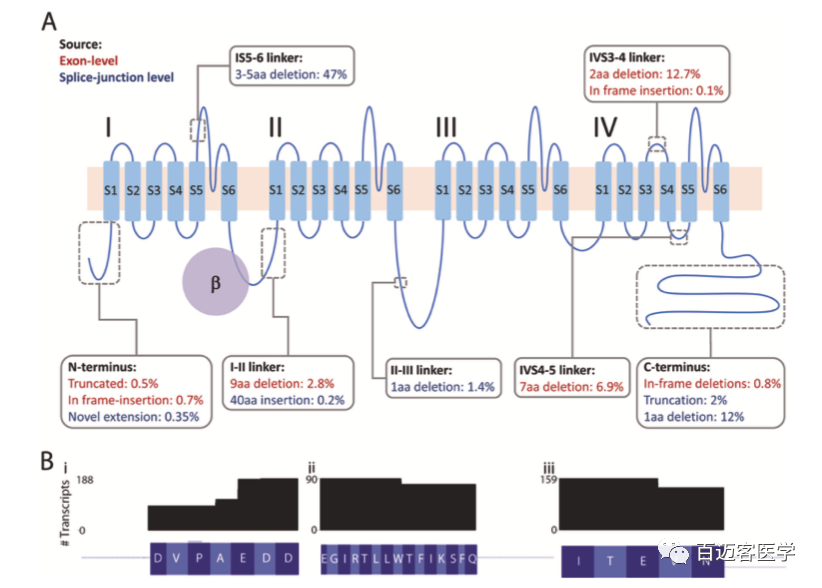
圖4
總結
長讀長測序技術的快速發展為準確獲得轉錄多樣性提供了可能,因為每一條read都包含一個完整的轉錄本。這對于具有復雜模型的基因尤其重要。由于CACNA1C剪接產生的CaV1.2蛋白對現有的鈣通道阻滯劑表現出不同的敏感性,因此有可能選擇性地針對疾病相關的CACNA1C亞型和/或那些在大腦與外周差異表達的CACNA1C亞型,提供既更有效又更無外周副作用的新型精神藥物。綜上,這些觀察結果證明了ONT長讀長測序對于準確描述轉錄本結構和選擇性剪接的重要性。
參考文獻:
Clark Michael B,Wrzesinski Tomasz,Garcia Aintzane B et al. Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain.[J] .Mol. Psychiatry, 2020, 25: 37-47.

]]>
ONT測序結果展示
作物類(部分)

林木類(部分)

動物(部分)

水產(部分)

中藥材(部分)
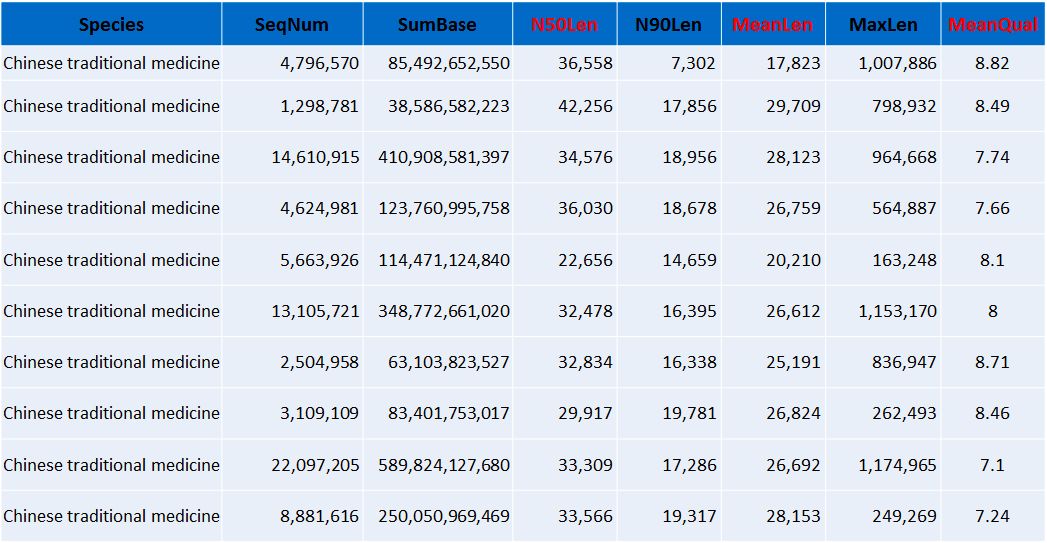
注:Species:分析的物種信息;SeqNum:各個長度范圍內序列的數目;SumBase:指各個長度范圍內序列的總長度;N50Len:reads N50長度;N90Len:readsN90長度;MeanLen:平均reads長度;MaxLen:最長reads長度;MeanQual:質量值。
以上是總結的部分作物類、林木類、動物、水產和中藥材的下機數據結果展示,從以上的數據不難看出,平均raeds長度幾乎均在20Kb以上,最長reads高達1.6Mb以上(不同樣品DNA抽提難易程度不同,會造成一定的影響)。
基因組組裝結果展示
上表中最后一列MeanQual就是下機數據的質量值,與堿基準確度的換算公式為:準確度 = 1-10^(-Q/10),經計算? Nanopore下機數據單堿基的平均準確率約為86%左右,這樣經過校正的數據再用Canu、SMARTdenovo、WTDBG等軟件進行基因組的組裝,再經過二代數據的polish之后,堿基的準確度可達到99.99%以上呢!
廢話少說,直接上組裝結果!
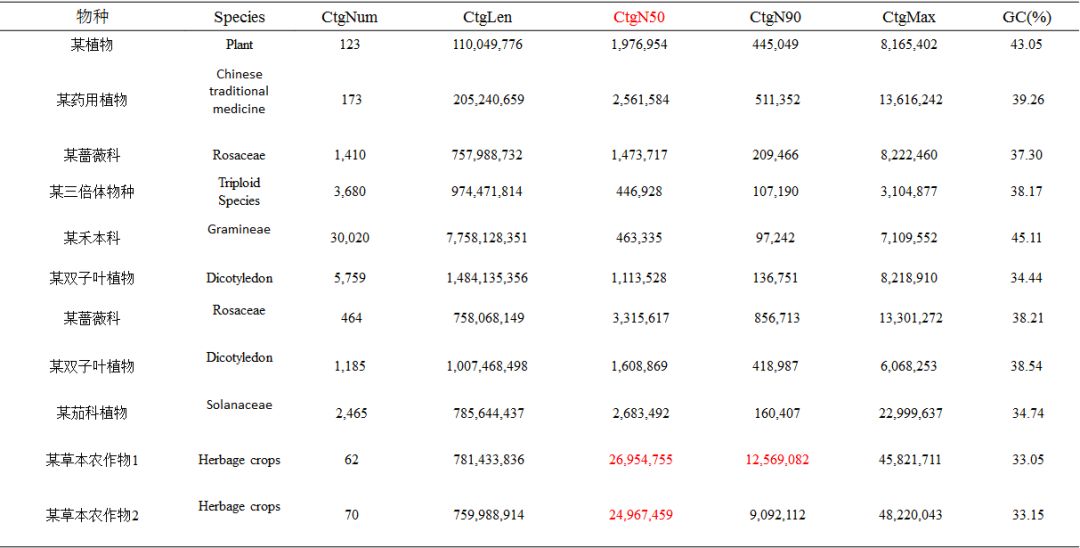
植物(部分)
動物(部分)
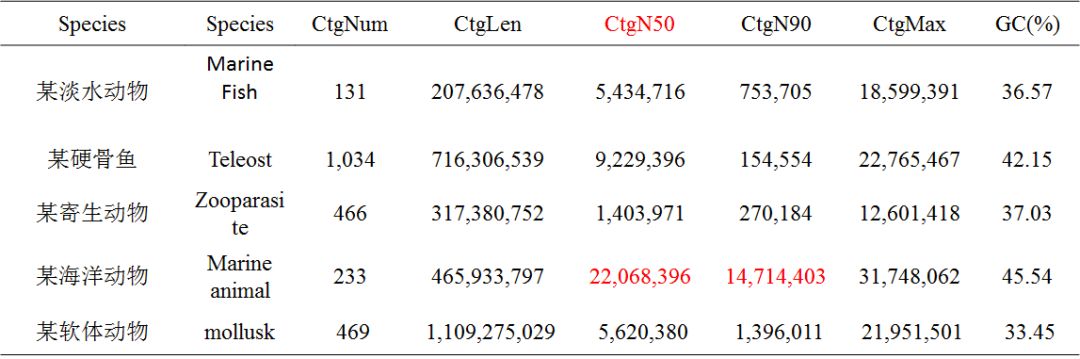
注:Species:分析的物種信息;CtgNum:contig數目;CtgLen:contig總長度;CtgN50:contigN50長度;CtgN90:contigN90長度;CtgMax:最長contig長度;GC(%):GC含量占比。
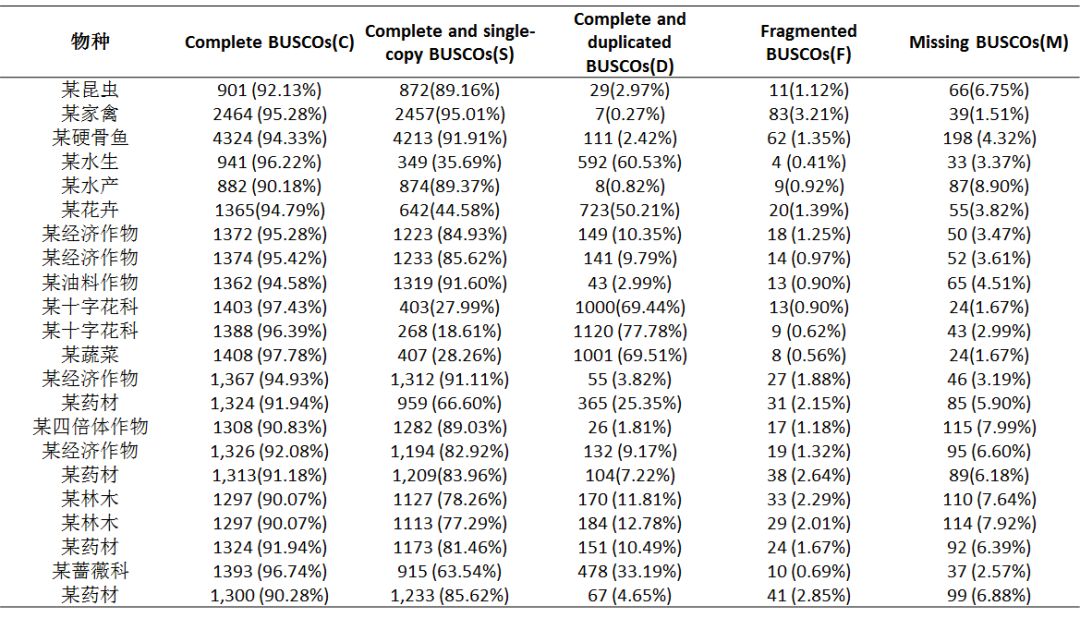
注:物種:分析的物種信息;Complete BUSCOs:找到完整基因數;Complete and single-copy BUSCOs:其中單拷貝基因數;Complete and duplicated BUSCOs:多拷貝基因數;Fragmented BUSCOs:預測不完整基因數;Missing BUSCOs:沒有預測出來的基因數。
評估結果顯示基因完整度均在90%以上!!說明Nanopore數據的組裝連續性和完整性都是非常好的,是值得廣大科研工作者信賴的哦!
百邁客ONT平臺發展歷程
?

第一代測序技術應用了Sanger雙脫氧鏈終止法,它的讀長可達1000bp,準確率高達99.999%,但測序前需要對特定區段進行引物設計且通量低,很難應用于組學方面的研究。基于此特點,涌現出二代測序技術,它主要的特點為短讀長,高通量。以Illumina?Solexa為例,它采用邊測序邊合成的方法,首先利用超聲波將DNA打斷成200-500bp小片段文庫,加接頭后DNA片段隨機附著于flowcell表面,經過橋式PCR擴增形成“DNA簇”,實現堿基信號強度放大,采用邊合成邊測序的方法,進行全基因組全面,準確的測序。

圖1? NovaSeq 6000
百邁客目前主要應用2017年Illumina平臺推出的NovaSeq系列測序平臺,雖然較于以往二代平臺,它的測序質量值、Index的測序識別、DNA文庫冗余度等指標有了明顯提升,但無法克服短讀長的reads 在基因組組裝、大片段變異檢測、轉錄組、甲基化等研究中的短板。基于此情況,三代測序應運而生。
目前,三代測序的主要代表為PicBio和Oxford Nanopore Technologies(ONT)這兩大測序平臺,以ONT平臺為例,它主要通過電信號識別堿基序列,單鏈DNA/RNA通過納米孔(蛋白通道),不同的堿基會形成特征性離子電流變化信號,通過對這些信號的檢測,得到堿基序列,完成測序。與二代相比,它主要的優勢在于在測序前,不會將DNA樣品打斷成小片段,而是對我們提取DNA進行片段篩選,一般篩選10-100kb大小的片段進行測序,這就對我們前期提取的DNA質量要求較高。
三代測序技術的出現,為復雜的多倍體基因組組裝帶來了福音。這種基因組由于倍性多,重復序列高,而二代測序局限于產生單倍體間的共有序列,導致此類物種的研究停滯不前。而ONT平臺由于其長讀長,跨越完整的重復區域,大的結構變異也得到了很好的檢測。eg. 納米孔測序技術可以將T-DNA結構的分辨率提升到36Kb。這就意味著,在這類突變體功能基因定位時,可以直接通過測序的方式,找到材料中T-DNA的插入位置及拷貝數,從而找到功能基因,實現基因克隆。和傳統的圖位克隆比較,將大大縮短定位周期。傳統的自然突變材料,如果已經有定位區段,應用二代檢測SNP,ONT檢測SV的方式可以讓我們的功能基因克隆方面事半功倍。
在基因組組裝方面,以生菜基因組為例,短讀長的二代測序組裝出21116個contig和2.21G的基因組,基于ONT平臺,則產生了1169個contig,contig N50為7.3Mb。二代數據產生了想較于三代數據18倍的contig用于基因組組裝,而三代平臺讀長的優勢為高質量的基因組組裝提供了便利。在轉錄組研究方面,ONT平臺的長讀長可以為我們帶來完整的轉錄異構體的序列,且可做定量研究,這將避免二代短片段數據在轉錄本組裝上的錯誤,更好的應用于轉錄組研究。

ONT做為新一代測序技術,已逐漸廣泛應用于科學研究中。百邁客一直致力于ONT平臺的探索與研發,目前擁有MinION、GridION X5、PromethION等多種3代測序平臺,且積累了豐富的項目經驗,期待你的加入哦~
如果您的科研項目有問題,歡迎點擊下方按鈕咨詢我們,我們將免費為您設計文章方案。

]]>
ONT測序技術在多個方面具有非常強悍的優勢,然而,一份合格的下機數據才是科研成功研究的基礎,為保證得到準確的轉錄組結構分析和定量結果,需要對測序數據進行嚴格的質控評估。那么我們今天一起學習一下《Summary statistics and QC tutorial》,ONT官方提供的對測序raw?data進行全面數據質控的教程。
介紹
此教程適用于指導對單個nanopore測序芯片產出的數據進行評估,評估的主要內容如下所示:
1、測序產出(測序得到多少reads,多大數據量);
2、測序數據的質量和長度分布;
3、如果加入了barcode序列進行混樣建庫,測序數據在不同樣品的分布。
準備
直接到教程的github頁面下載或通過git命令下載:
git clone https://github.com/nanoporetech/ont_tutorial_basicqc.git QCTutorial
后續分析會用到下載目錄QCTutorial下的以下內容:
1) Nanopore_SumStatQC_Tutorial.Rmd:Rmarkdown文件,說明文檔和用于執行分析。
2) RawData/lambda_sequencing_summary.txt.bz2:示例文件,Guppy對測序reads進行堿基識別生成的相關信息文件。
3) RawData/lambda_barcoding_summary.txt.bz2:示例文件,用于區分混樣建庫時多樣品的barcode信息。
4) environment.yaml:指定分析所需軟件包及計算環境的文本文檔。
5) config.yaml:配置文件,用于指定分析所需的輸入。
2、創建Conda環境
為了方便執行分析所需軟件包及其依賴的安裝及管理,需要安裝Conda并創建用于此分析的環境。
1)?Conda安裝(Python3版本的Miniconda):
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
bash
2)?創建Conda環境及環境激活(第1步中下載的environmen.yaml用于環境初始化):
創建環境:conda env create –name BasicQC –file environment.yaml
激活環境:source activate BasicQC
分析
進行分析之前需先準備配置文件,通過修改準備步驟下載的config.yaml中相應的參數來完成,需要修改的內容主要有:
| 修改內容 | 內容說明 | 示例 |
|---|---|---|
| inputFile | 堿基識別的統計信息 | sequencing_summary.txt.bz2 |
| barcodeFile | 混樣建庫的barcode信息 | barcoding_summary.txt.bz2 |
| basecaller | 堿基識別工具 | Guppy 2.1.3 |
| flowcellId | 測序芯片ID | FAK41706 |
注:如為單樣品測序無barcode信息,則barcodeFile部分為空。
準備完成后,可以通過命令行啟動分析,命令如下:
R –slave -e ‘rmarkdown::render(“Nanopore_SumStatQC_Tutorial.Rmd”, “html_document”)’
如果習慣圖形界面操作,也可以通過Rstudio載入Rmarkdown文件執行分析:
結果
上述分析完成后會將分析結果存放至HTML文件,可用瀏覽器打開Nanopore_SumStatQC_Tutorial.html進行查看。對單個芯片約1M reads分析的部分結果展示如下(結果來自教程,堿基識別使用Guppy 2.1.3,根據識別序列的平均質量值將其分為pass和fail兩種,質量值閾值默認為7):
1、總結
展示了數據產出的總體情況(如下圖,本分析中堿基識別共產出991,715條序列,14.6G堿基)。

2、質量長度
此部分展示了對識別出的所有序列質量和長度信息的統計結果,包括序列的平均長度,N50和平均質量,序列長度和質量的密度分布等

3、測序表現
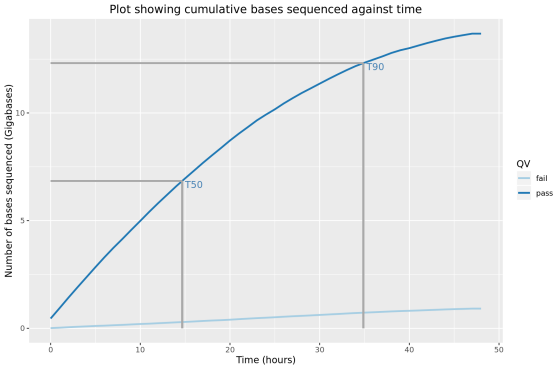
此部分內容統計了隨測序時間變化,測序累計序列個數,堿基個數,測序速度和有效工作納米孔數等指標的變化情況。

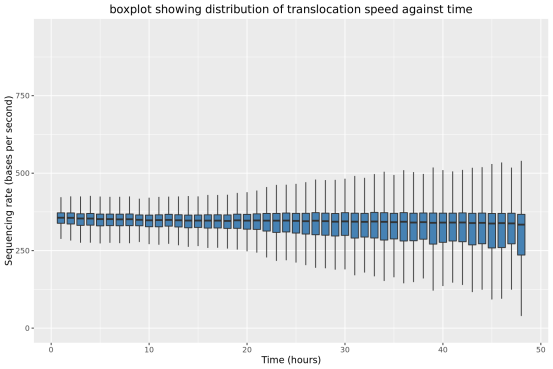
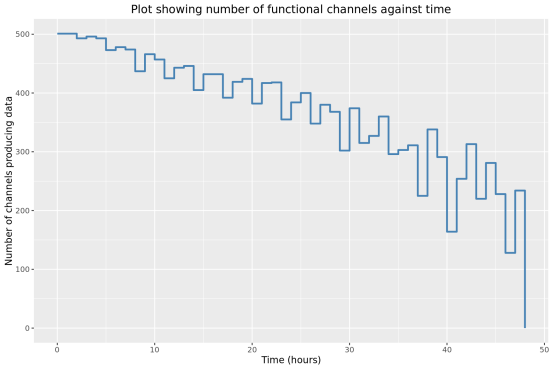
4、區分混樣
在加入barcode序列混樣測序的情況下,barcode識別區分的結果展示如下,包括barcode識別效率,區分的文庫個數及每個文庫中序列個數占比和長度信息等。

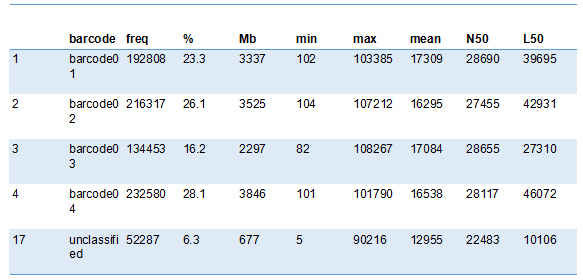
上面展示了分析結果的部分內容,更多細節的內容可參考底部的相關鏈接。
rawdata的質控評估只是整個信息分析的開始,是為了對測序數據有大致的整體認識,以便更好地指導后續分析。然而分析的每個環節都會對最終結果產生影響,因此每一步的處理都要深思熟慮。
小編寄語
2018年8月牛津納米孔公司與百邁客公司達成長期合作,擁有MinION、GridION X5和PromethION三種型號全套納米孔測序儀。至今已積累了豐富的項目經驗,全長轉錄組成功案例先后發表在《Plant Biotechnol J》、《J Hazard Mater》、《Biotechnol Biofuels》、《Sci Rep》、《Fish & Shellfish Immunology》等國際知名期刊,已發表文章研究物種分別有楊樹、吳松草、風箏果、甘薯、野生甘薯、兔子、跳甲、花羔紅點鮭和辣椒,覆蓋領域分別為林木、哺乳動物、昆蟲、水產和作物等。
如您有任何全長轉錄組等相關問題,歡迎點擊下方按鈕,我們將竭盡全力為您答疑、設計方案和提供高分成功案例等。
參考鏈接:
https@//github.com/nanoporetech/ont_tutorial_basicqc(@換成:)
https@//community.nanoporetech.com/knowledge/bioinformatics(@換成:)
]]>