英文名:?Integration of full-length transcriptomics and?targeted metabolomics to identify?benzylisoquinoline alkaloid biosynthetic genes in?Corydalis yanhusuo
雜志:Horticulture Research
影響因子:5.404
研究背景
延胡索( Corydalis yanhusuo W.T. Wang) ,別名元胡,罌粟科紫堇屬多年生草本植物,常以其干燥塊莖入藥,是世界上具有低成癮性和耐受性的鎮痛中藥,鎮痛效價約為嗎啡的60%。四氫巴馬汀和左旋紫堇達明已被確認為延胡索中具有鎮痛活性成分,可作為阿片類鎮痛藥的替代品,但含量低下,產量較小的缺點制約著該類藥物的應用。
研究目的
對合成原小檗堿型芐基異喹啉生物堿的相關基因進行了挖掘,為后期利用合成生物學與植物代謝工程生產延胡索中具有鎮痛效果的痕量化合物奠定了基礎。
材料方法
延胡索,成熟期的葉和塊莖,道地產區浙江磐安
代謝:UPLC-Q-TOFMS定性定量
轉錄組:二代+三代全長轉錄組測序
研究結果
1、轉錄組和代謝組測序分析
該研究以來自道地產區浙江磐安的延胡索成熟期的葉和塊莖為研究對象,采用UPLC-Q-TOFMS定性定量分析了延胡索中具有鎮痛活性的成分,并通過二代校準的三代全長轉錄組測序的方法對合成原小檗堿型芐基異喹啉生物堿的相關基因進行了挖掘。
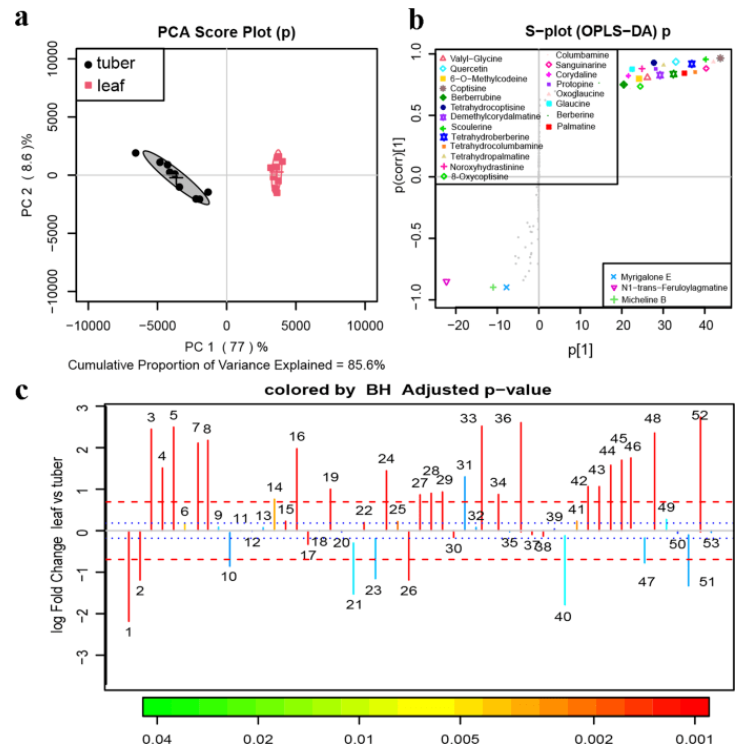
延胡索提取物的塊莖和葉組QTOF-MS數據的代謝組學多元分析
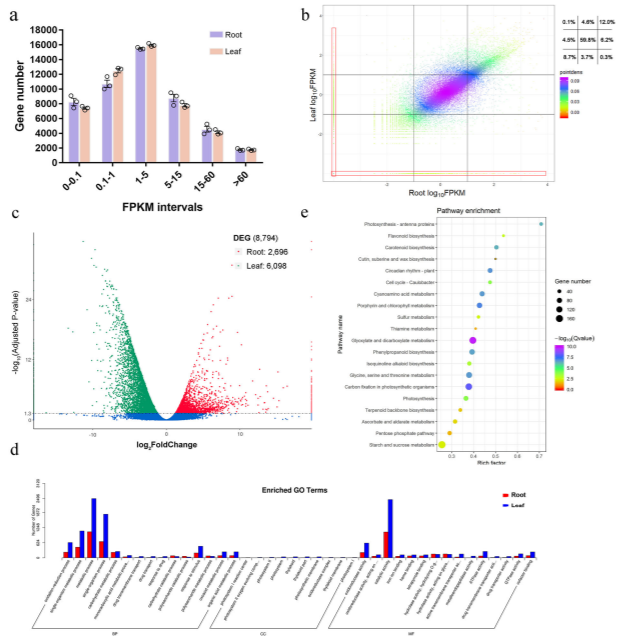
延胡索塊莖和葉片之間基因的差異表達
2.、轉錄組-代謝組聯合分析
對處于不同器官的組織樣品進行了轉錄組-代謝組聯合分析,最終鑒定到了101個參與芐基異喹啉生物堿(benzylisoquinoline alkaloid,BIA)生物合成途徑的unigenes和38種在延胡索葉與塊莖中含量具有顯著差異的代謝物,并對其中19種典型的代謝物進行了器官差異性豐度測定。結果顯示,目前已知的合成途徑在延胡索中報導過的BIAs合成途徑中均成功對應到至少一種關鍵合成酶的unigene,說明BIA的空間分布差異主要受到轉錄水平上的調控。
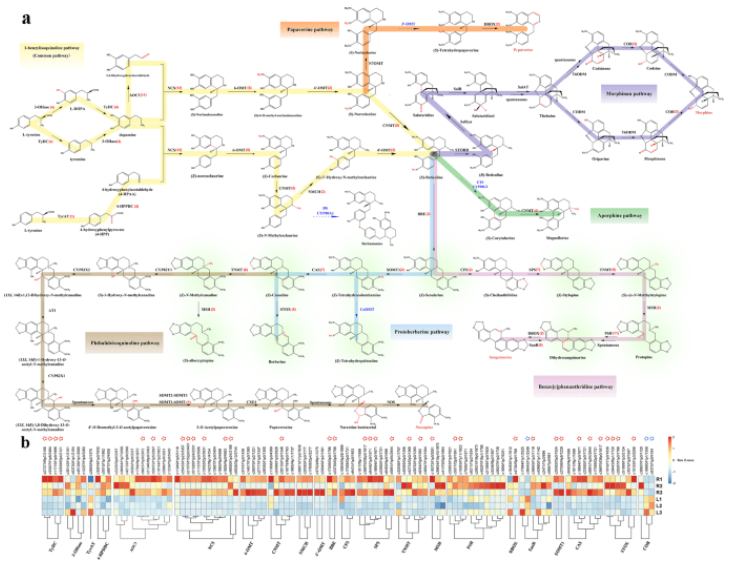
芐基異喹啉生物合成途徑
3、 OMT蛋白家族系統發育樹分析
進一步研究發現,其中參與合成具有廣泛臨床鎮痛潛力的關鍵代謝物四氫巴馬汀的酶可能與唯一已知的黃連中負責催化這一步反應的ColumbamineO-methyltransferase(CoOMT)存在較大的蛋白質序列差異。通過系統發生樹分析,作者們推斷出至少存在10種unigenes的翻譯產物可能催化四氫巴馬汀的合成。 特異性的氧甲基化是四氫巴馬汀與紫堇達明合成的關鍵,延胡索中是否存在特殊的一類OMT或者具有不同底物特異性的OMT則是接下來將要重點解析的重點。
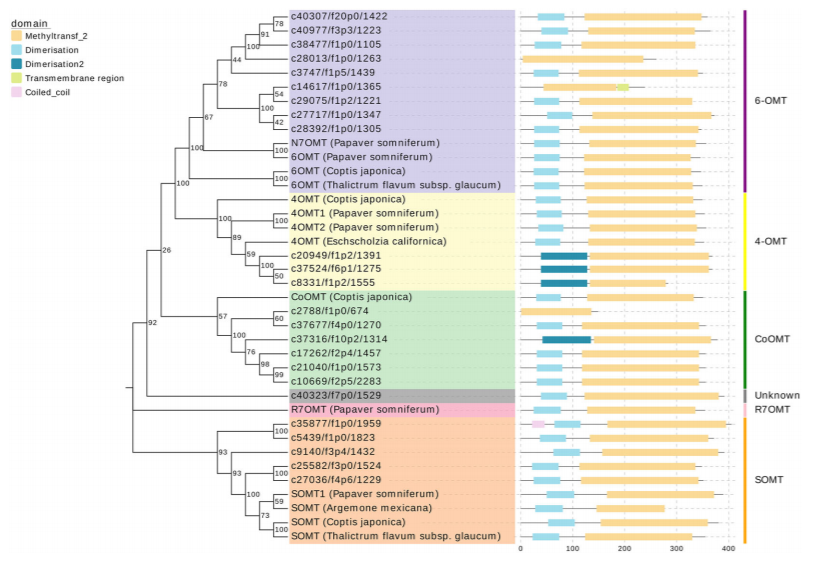
OMT蛋白家族系統發育樹
文章亮點
本研究明確了鎮痛成分的生物合成機制,同時為進一步生化水平上的活性驗證提供了方向,且為后期延胡索功能基因組的解析奠定基礎。
?
]]>英文名:Multi-omics reveals functional genomic and metabolic mechanisms of milk production and quality in dairy cows
雜志:Bioinformatics,2020
影響因子:5.61
研究背景
提高人類不可食用的農作物副產品的利用率是維持畜牧業可持續性的當務之急,在過去的幾十年中,已經做出了許多努力來利用農作物副產品作為飼喂奶牛的飼料來源。但是,與高質量和高成本的牧草苜蓿干草(AH)相比,大多數農作物副產品,例如玉米秸稈(CS,是玉米生產的副產品),營養價值較低,通常會降低牛奶產量和品質。許多研究已經使用化學分析來評估這些飼料的營養成分,消化,利用率,微生物發酵的差異及其對牛乳生產的影響。最近,基于組學的技術也已被用于了解飼喂CS日糧的母牛在分子水平上的生理和生物學變化。但是,牛奶的生產過程可能會受到多個器官/組織的影響和調節。對不同器官中復雜的牛奶生產生物學過程的總體調節機制缺乏全面的了解,無法獲得更可實現的結果。
研究目的
提高反芻動物對人類不可食用的農作物副產品的利用,以生產供人類消費的優質牛奶是一項新興的全球性任務。我們進行了一項基于多組學的研究,以了解奶牛飼喂低品質農作物副產品時牛奶生產的調控生物學過程,旨在提高其利用率。
材料方法
1、實驗樣本:
(1)養殖條件:16頭中國荷斯坦奶牛(產奶量=29.4±2.16kg/d;日產奶量=164±27.5d;均價=3.6±1.8;平均±SD)。根據牛奶產量,在隨機分組設計中將其分配給以下2種處理方法之一:苜蓿干草為主的日糧(AH,n=8)和玉米秸稈基礎飲食(CS,n=8)。AH和CS日糧含有52.9%和54.3%的干物質(DM),16.7%和16.2%的粗蛋白(以DM為基準),31.1%和36.3%的中性洗滌劑纖維(以DM為基準),18.5%和19.5%的酸性洗滌劑纖維(以DM為基礎),非纖維狀碳水化合物(NFC,DM為基礎)分別占40.6%和36%。每天在06:30、14:00和20:00h隨意飼喂3次,每次3至5%的飼料,使用混合飼料。
所有母牛都被喂食65天以收集生物流體樣本(瘤胃液,血清,牛奶和尿液)。在這項研究中測量了這16頭母牛的血液參數,瘤胃氨氮和揮發性脂肪酸。其中,考慮到動物屠宰和多組學測量的成本,按照公開的方法從每組中隨機選擇6頭母牛進行屠宰后再喂養25天以收集肝臟和MG組織。使用SAS的PROCTTEST(版本9.4)分析瘤胃發酵參數,血液參數,瘤胃微生物多樣性和功能數據。
(2)樣本收集:收集飼喂65天的16頭牛生物流體樣本(瘤胃液,血清,牛奶和尿液)和血液;再喂養25天后從每組中隨機選擇6頭母牛進行屠宰以收集肝臟和MG組織。測量16頭母牛的血液參數,瘤胃氨氮和揮發性脂肪酸。
2、實驗方法:
(1)代謝組學:肝臟和MG組織,GC-TOF/MS。
(2)宏基因組:瘤胃微生物組,IlluminaMiSeq。
(3)轉錄組:肝臟和MG組織轉錄組分析。
研究結果
1、飼喂基于AH和CS的日糧的奶牛之間的表型變化
與優質飼草(苜蓿干草)相比,以低品質CS喂養母牛時,觀察到的牛奶產量,牛奶蛋白,乳糖和牛奶效率顯著降低。對血液中葡萄糖濃度的后續分析顯示,CS組的血糖濃度顯著低于AH組(P=0.03)(表1)。在飼喂CS的牛的瘤胃中,乙酸與丙酸酯的比例和氨氮的濃度均顯著較高(P<0.01)(表1)。在CS喂養的動物中,乙酸鹽和丙酸鹽的瘤胃摩爾比例分別顯著較高和較低(P<0.01)(表1)。

Table 1. The blood and rumen fermentation parameters in the dairy cows fed with alfalfa hay and corn stover based diets.
2、根據宏基因組學,AH組和CS組之間瘤胃微生物組的組成和功能差異
從瘤胃微生物群中鑒定出總共784屬,包括古細菌,細菌和真核生物,在所有動物中共檢測到其中的111屬。進一步的比較表明,變形桿菌,纖維桿菌和門菌(圖1a)和擬桿菌,副細菌,纖維桿菌,卟啉單胞菌,Paludibacter和Victivallis屬的相對豐度顯著較高(P<0.05),而在飼喂CS的奶牛的瘤胃中,密螺旋體屬和琥珀酸單胞菌屬明顯降低(P<0.05,相對豐度>0.02%)(圖1b)。在396種優勢細菌中(相對豐度>0.01%,補充表2),麥芽螺旋體僅在AH組的瘤胃中發現,而在CS喂養的牛瘤胃中,T.saccharophilumandT.succinifaciens的相對豐度顯著降低(P<0.05)。在兩組奶牛的屬或種水平上,古細菌和真核生物的豐度均未發現差異。宏基因組功能分析揭示了兩組在1級基因功能中碳水化合物代謝的相對相對豐度不同(圖1c,P=0.032)。碳水化合物代謝中第4級功能的比較表明,編碼乳醛還原酶(EC1.1.1.77),I型谷氨酰胺合成酶(EC6.3.1.2),甲基丙二酰輔酶A脫羧酶(EC4.1.1.41),琥珀酸的基因相對豐富CS組的脫氫酶(EC1.3.5.1)和α-木糖苷ABC轉運蛋白顯著降低(圖1d,P<0.05)。
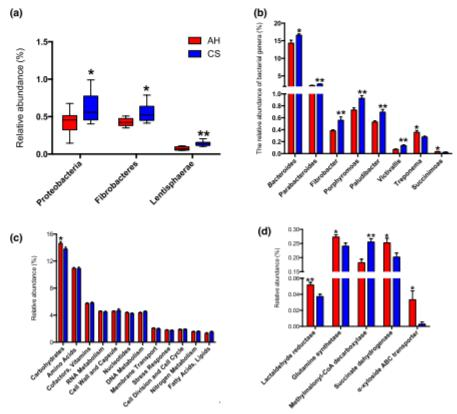
Figure 1. Rumen microbial difference between AH and CS groups
3、基于代謝組學和代謝組學綜合分析的AH和CS組瘤胃微生物代謝特征差異
瘤胃液中55種明顯不同的代謝產物中,CS喂養的動物中49種的代謝產物含量較低,通過將微生物分類群相對豐度,碳水化合物代謝中的基因豐度和17種高度豐富的微生物代謝物相關聯,研究了微生物代謝物與微生物之間的關系。核心宏基因組相關元素包括三個細菌屬(纖維桿菌,琥珀酸桿菌和螺旋體)),三種細菌種類(解淀粉琥珀酸桿菌,琥珀酸梅毒螺旋體和嗜糖鏈球菌)和三種微生物功能(甲基丙二酰輔酶A脫羧酶,琥珀酸脫氫酶和乳醛還原酶)(圖2a)。琥珀酸脫氫酶基因和解淀粉鏈球菌的相對豐度分別與16種代謝物(r介于0.43至0.68,P<0.1)和15種代謝物(r介于0.43至0.76,P<0.1)呈正相關。對上述瘤胃代謝組和代謝組的綜合分析顯示,當給動物喂食CS時,瘤胃中產生較低的丙酸根和較高的乙酸鹽的潛在機理(圖2b)。
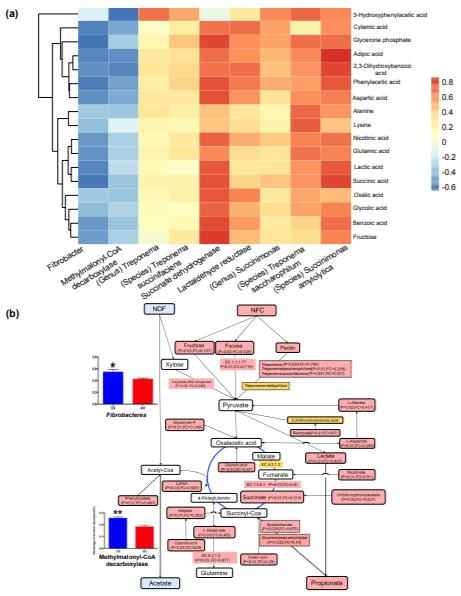
Figure 2. Rumen microbial metabolic signature difference between AH and CS groups based on the metagenome-metabolome integrated analysis.
4、菌體和屬水平的微生物分類學分析
代謝物譜分析分別在肝臟和MG組織中鑒定出270和273種代謝產物(重疊179種)。肝臟中28種代謝產物的豐度差異顯著(VIP>1&P<0.05);在CS喂養的動物中,其中15個較低,而13個則較高(VIP>1&P<0.05)。在飼喂CS的動物中,MG組織中3種代謝物含量較低,而6種代謝物含量較高(VIP>1&P<0.05)。圖3顯示了ROC曲線分析的肝臟和MG組織中AUC最高的前3種代謝產物及其在AH和CS組中的相對濃度。肝臟中的馬尿酸(HCA,AUC=1,log2FC=7.54;圖3a),亮氨酸(AUC=1,log2FC=11.01;圖3b),胱氨酸(AUC=0.972,log2FC=-4.24;圖3c)表現出最*的預測性能,可以區分AH和CS。
MG中的馬來酸(AUC=0.972,log2FC=-2.88;圖3d),鄰苯三酚(AUC=0.833,log2FC=6.26;圖3e)和琥珀酸(AUC=0.833,log2FC=-3.01;圖3f)是分離AH和CS動物的潛在生物標記。通過IPA識別,CS喂養的奶牛的肝臟中關鍵顯著下調的途徑是氨基酸(AA)代謝(P=1.66E-07,Z評分=-3.12),糖異生(P=3.86E-08,Z評分=-2.61))以及肝臟中的維生素和礦物質代謝(P=1.47E-15,Z分數=-2.42)(圖3g);MG中AAs的攝取(P=1.49E-08,Z分數=-2.77),攝取L-AA(P=4.52E-07,Z分數=-2.41),碳水化合物代謝(P=2.10E-11,Z分數=-2.24)和葡萄糖-6-磷酸的氧化(P=1.01E)-11,Zscore=-2.21)途徑顯著下調。(圖3h)
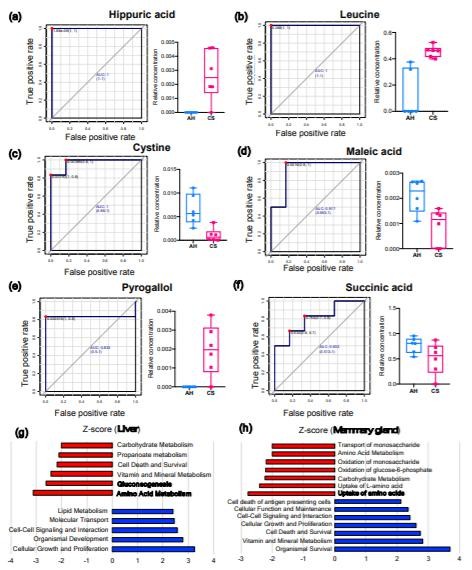
Figure 3. The biomarker and functional analysis in the liver and mammary gland tissues between AH and CS groups.
5、AH和CS組之間差異表達的基因和功能分析
分別從肝臟和MG轉錄組中分別獲得22.63±1.95和20.03±271萬原始序列讀數。基因表達的密度在每個組織內的飲食處理之間沒有顯示出明顯的差異,但是在兩個組織之間存在顯著的差異。在AH和CS組之間,總共在肝臟中發現了67個基因(CS組中9個上調和58個下調)DE基因(|倍數變化|>2&FDR<0.05)(圖4a)。在CS奶牛的MG中,兩個基因(IGFBP1和ENSBTAG00000047957)以及六個基因(包括PDZK1IP1,LALBA,CSN1S2,LOC505033,CSN3和PDPN)分別顯著上調和下調(圖4b)。基于DE基因,在AH和CS組之間分別在肝臟和MG組織中鑒定出165個和19個功能性GO術語。

Figue 4. The differential expressed genes between AH and CS groups and coexpressed gene module-biomarker correlation analysis in the liver and mammary gland tissues.
6、代謝機制的確定作為營養物質分配的結果
在兩個動物組之間,AA代謝途徑是肝臟中最顯著不同的途徑(圖5a)。對AA代謝途徑的進一步影響分析表明,亞途徑,甘氨酸,絲氨酸和蘇氨酸代謝的影響較大(圖5b,P=0.029,影響值=0.597)。因此,我們對肝臟中與甘氨酸相關的反應進行了徹底的分析,并研究了它們與體內體液中代謝物的關系。綜合比較表明,苯甲酸,糖胺(GAA)和HCA在整個人體的AA代謝中起著關鍵作用,當母牛飼喂CS時,隨后會影響牛奶的合成(圖5c)。用CS喂養的奶牛肝臟中甘氨酸和HCA的濃度更高,進一步支持了我們的推測,即瘤胃(微生物)和肝臟(宿主)中的苯甲酸代謝都可能受到影響(圖5c)。對肝,血清和尿液進行的HCA生物標志物分析顯示出一致的結果(圖5d),這進一步表明,從肝臟到尿液的HCA循環是CS下牛奶產量變化的生物標志物。
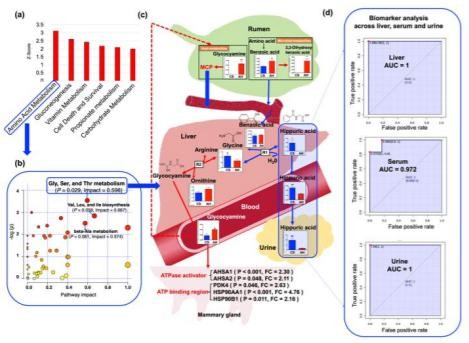
Figure 5. The roles of systematic metabolites across multiple organs and biofluids when cows fed corn stover 。
文章創新點
這項研究揭示了低質飼草飲食下跨不同生物流體和組織的與牛奶生產相關的生物學機制,這為未來作物副產品的利用和可持續反芻動物的生產提供了新穎的理解和潛在的改善策略。總之,對瘤胃,肝臟和MG組織中不同分子(DNA,RNA,代謝物)的多組學評估表明,當給母牛喂食CS時,乳牛生產中所涉及的代謝和分子生物學機制。對高品質和低品質飼料來源的奶牛進行系統的比較,為提高人類不可食用的飼料用于牛奶生產的生物利用度提供了基本的了解。已發現馬尿酸是導致牛奶產量低的代謝生物標志物,暗示了與低質飼草利用代謝機制有關的未來評估參數,在開發用于繁殖和選擇或分類動物的工具以優化農場管理方面,這是有希望的。
]]>
轉錄組+代謝組數據挖掘實現思路
轉錄組代謝組聯合分析一、轉錄組云平臺個性化可視化交互
整體層面分析樣本間/組間差異:PCA分析、相關性分析、WGCNA分析、聚類熱圖等
差異層面分析比較組差異基因:
1、差異基因篩選: 差異火山圖,聚類熱圖、韋恩圖、WGCNA、共表達趨勢(K-Means);
2、差異功能基因挖掘:差異基因功能注釋和富集分析(COG、GO、KEGG等)、GSEA(有生物意義,無顯著性)、蛋白互作、轉錄因子預測
此外,百邁客提供個性化可視化交互—50+個性化功能+圖片交互(字體及配色調整、布局變換、數據篩選等),數據挖掘一網打盡;一站式科研服務—操作簡單,85%可視化交互覆蓋率,分析不限時不限次;光速周期—常規分析1min,分析結果極速可得!
轉錄組代謝組聯合分析二、代謝組云平臺個性化全面覆蓋
整體層面分析樣本間/組間差異:PCA分析、相關性分析、OPLS-DA分析、WGCNA分析、Kmeans聚類分析(代謝物)等
差異層面分析比較組差異代謝物:
1、差異代謝物篩選:差異火山圖,聚類熱圖、韋恩圖;
2、差異代謝物功能挖掘:差異代謝物能注釋和富集分析(COG、KEGG)、富集弦圖、ipath;
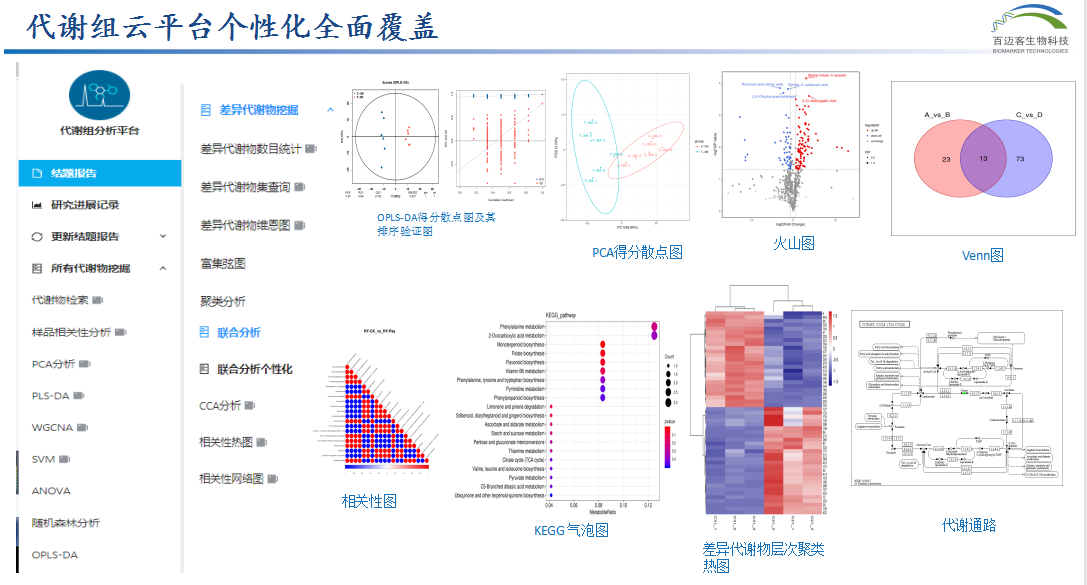
轉錄組代謝組聯合分析三、代謝組云平臺—多種聯合分析任你搭配
整體層面分析轉錄組與代謝組數據的相關性:PCA分析、O2PLS分析、WGCNA
差異層面獲得共表達的差異基因和差異代謝物:
1、差異基因和差異代謝物KEGG通路分析:KEGG通路比較分析(韋恩圖,富集)、KEGG通路可視化、Ipath 整合分析
2、、轉錄組和代謝組相關性分析:九象限圖、相關性熱圖和玄圖、相關性網絡熱圖、相關性網絡圖、典型相關分析(CCA)。
轉錄組代謝組聯合分析案例分享
中文標題:多組學聯合解析突尼斯軟籽石榴揮發性風味物質生物合成機制
英文標題:Transcription profile analysis for biosynthesis of flavor volatiles of Tunisian soft-seed pomegranate arils
期刊(IF):Food Research International
影響因子:7.425(2022年4月)
研究策略:轉錄組學+非靶代謝組(GC-MS)+生理指標測定
Doi:10.1016/j.foodres.2022.111304
研究方案
材料:采集來自云南大理市(Y_DTN)、云南麗江市(Y_LTN)、云南建水縣(Y_JTN)、云南曲靖市(Y_QTN)、四川會理市(S_DHT)和攀枝花市(S_PTN)以及河南滎陽市(H_HYT)共7個不同產區的石榴果肉樣品。
方法:RNA-seq;非靶代謝組;生理指標測定:pH值、可滴定酸度(TA)、總可溶性固體(TSS)和TSS/TA值
研究內容
1、生理指標+代謝組學分析:
7個產區突尼斯軟籽石榴揮發性化合物的熱圖:風味譜、糖、有機酸和維生素c含量存在顯著差異。所有石榴花粒中共鑒定出40種揮發性化合物,其中醇、醛、酸和二戊烯是影響石榴香氣品質主要因素。

揮發性化合物的熱圖
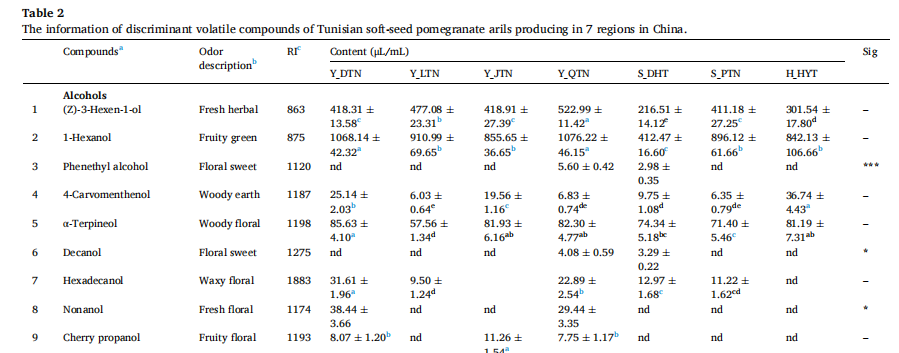
揮發性化合物鑒定(部分結果)
2、轉錄組分析:
WGCNA分析:1808基因(生物合成相關基因),1640個基因被分為11個不同的模塊,紅色、黃色、綠松石、藍色、品紅、綠色模塊與大多數揮發性化合物呈較高的正相關關系(CC > 0.5),表明這些模塊中的基因主要與所有石榴粥樣品中風味化合物的產生有關。
GO和KEGG富集;1808基因主要富集于分子功能的“裂解酶活性”,細胞成分的“葉綠體基質”,生物過程的“小分子生物合成過程”。鑒定了40條顯著富集的KEGG途徑,其中碳代謝生物合成的DGE數量最多。與其他次生代謝產物相關的途徑也被富集,包括淀粉和蔗糖代謝、糖酵解/糖異生等,這些富集的基因大部分屬于黃色、綠松石色和藍色模塊。這也表明,這些模塊中的基因的功能與KEGG富集分析相一致,而KEGG富集分析主要與初級和次生代謝產物的合成和代謝有關。
基因相關網絡:三個模塊與揮發性化合物的合成具有高度的相關性,但三個模塊中的基因表現出不同的組成和表達模式,說明不同模塊中的基因在揮發性化合物的合成中可能發揮不同的功能。

3、轉錄組+代謝組聯合:
相關性網絡圖:己酸和1-己醇是石榴果實中的關鍵揮發性化合物,分別與38個和31個DEGs顯著相關,例如Para-α-二甲基苯乙烯、2-非丙酮、肉豆蔻酸、苯乙烯和乙酸與不少于5個DGE具有顯著的高相關性。結果表明,植物的次級代謝途徑不僅僅是相應途徑中酶與底物反應的結果,還受到其他途徑中基因編碼蛋白與代謝物相互作用的影響。
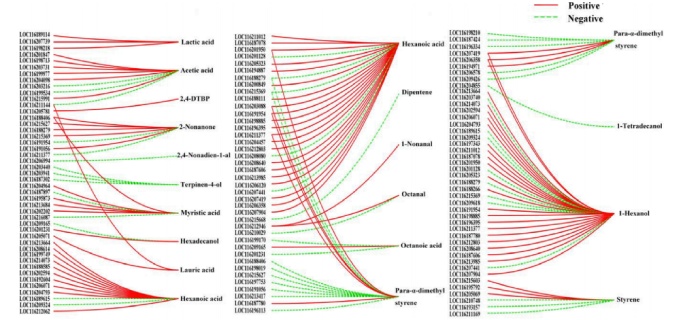
差異基因和差異代謝物相關性網絡圖(|r|≥0.8,p < 0.05)
]]>
多組學解析胰腺癌中結構變異與三維基因組的動態互作
Dynamic Interplay between Structural Variations and 3D Genome Organization in Pancreatic Cancer
發表雜志:Advanced Science
影響因子:17.521
發表日期:202205
發表單位:國家癌癥中心、中國醫學科學院腫瘤醫院和北京協和醫學院

研究背景
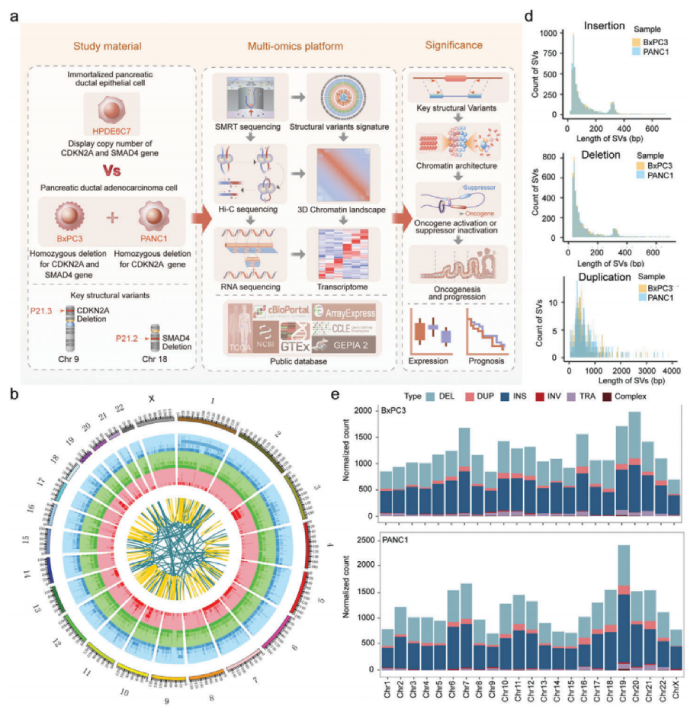
結構變異(SV)是基因組變異的來源之一,并可能導致癌基因生成。然而,人類癌癥中SV的識別和解釋在技術上仍然具有挑戰性。利用長讀長測序和Hi-C優勢檢測人類胰腺導管上皮癌發生結構變化的特征,揭示了3D染色質架構的廣泛重新編程,對闡明胰腺癌基因組結構變化特征對其發生發展的分子機制至關重要。
研究設計
材料:人類胰腺導管上皮細胞(HPDE6-C7)和人類胰腺癌細胞系PANC1和BxPC3;CCLE+GSE97003數據庫中的三條細胞系進行了轉錄組聚類分析
方法:Hi-C互作、單分子實時(SMRT)測序、RNA-seq、ChIP-seq(NCBI PRJEB27863)
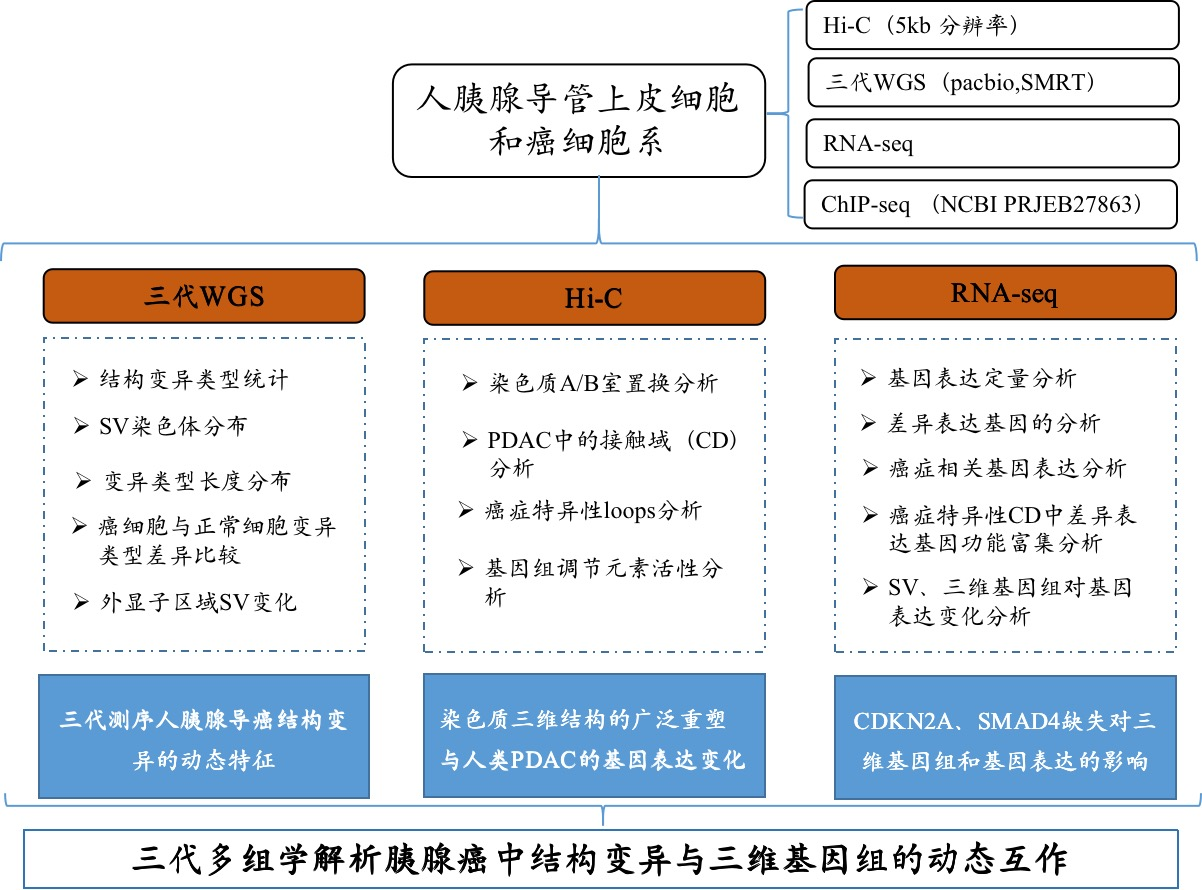
研究結論
研究人員通過整合三代人重測序、高通量染色質構象捕獲( Hi-C) 和 RNA-seq等多組學技術,對源自人胰腺導管上皮的胰腺癌細胞和正常人胰腺導管上皮細胞進行了深入研究和綜合分析,成功揭示了PDAC細胞中SV的特征圖譜和染色體空間結構的多尺度重塑,并進一步闡釋了SV與染色質三維結構之間的復雜的相互作用及其對基因表達的影響。這一發現為全面了解SV在胰腺癌發展中的致病機制提供了全基因組資源和新的空間視角,這可能有助于識別新的預后標志物和治療靶點。
文章亮點
借助三代人全基因組重測序技術,揭示胰腺癌關鍵驅動基因CDKN2A和SMAD4的復雜基因組重排,并且結合Hi-C技術識別了染色質空間構象的改變,并從線性角度和3D角度進一步闡明了它們對癌基MIR31HG、MYO5B等表達的影響,這是首次采用多組學策略集成了長讀長測序和Hi-C技術研究結構變異(SV)對胰腺癌三維基因組的影響,為PDAC診治開發提供了遺傳和分子基礎的根本新見解。
研究結果
1、人類胰腺癌結構變異的特征
利用SMRT測序長度長的優勢全面研究了正常胰腺導管上皮細胞惡性轉化過程中發生的SV的動態譜,檢測到PANC1、BxPC3和HPDE6C7中存在大量的結構變異(SV),分別為20 805、21 168和23 035個SV。兩種常見的SV類型是插入和缺失,分別占所有SV的約50%和41%。值得注意的是,與正常上皮細胞系相比,癌癥細胞系中兩種以上簡單SV斷端重疊的復雜SV數量增加了兩到四倍,這表明在惡性轉化過程中,基因組不穩定性大大增加。對這些SV進行染色體區域分布、長度分布分別進行分析,插入、缺失和重復表現出相似的特征,大多數長度都在1kb以內,表現出細胞異質性。
人胰腺導管癌SV整體景觀
研究了在其外顯子區域受到SV直接影響的基因,并分別在PANC1、BxPC3和HPDE6C7中檢測到了1017、1061和1683個基因的變化。在PANC1中獨立檢測到14號染色體上的DICER1擴增,19號染色體上的AKT2缺失和20號染色體上的GNAS插入。在BxPC3中特異性地檢測到9號染色體上的TNC缺失,11號染色體上的RRAS2插入和18號染色體上的SMAD4缺失。BxPC3樣本中還存在許多基因大片段缺失,長度超過1.7 Mb,包括CDKN2A、CDKN2B、MTAP和DMRTA1等基因。基于長讀長測序結果建立了人類胰腺癌中SV的特征,為全面研究SV在這種致命惡性腫瘤中的發病機制提供寶貴的資源。
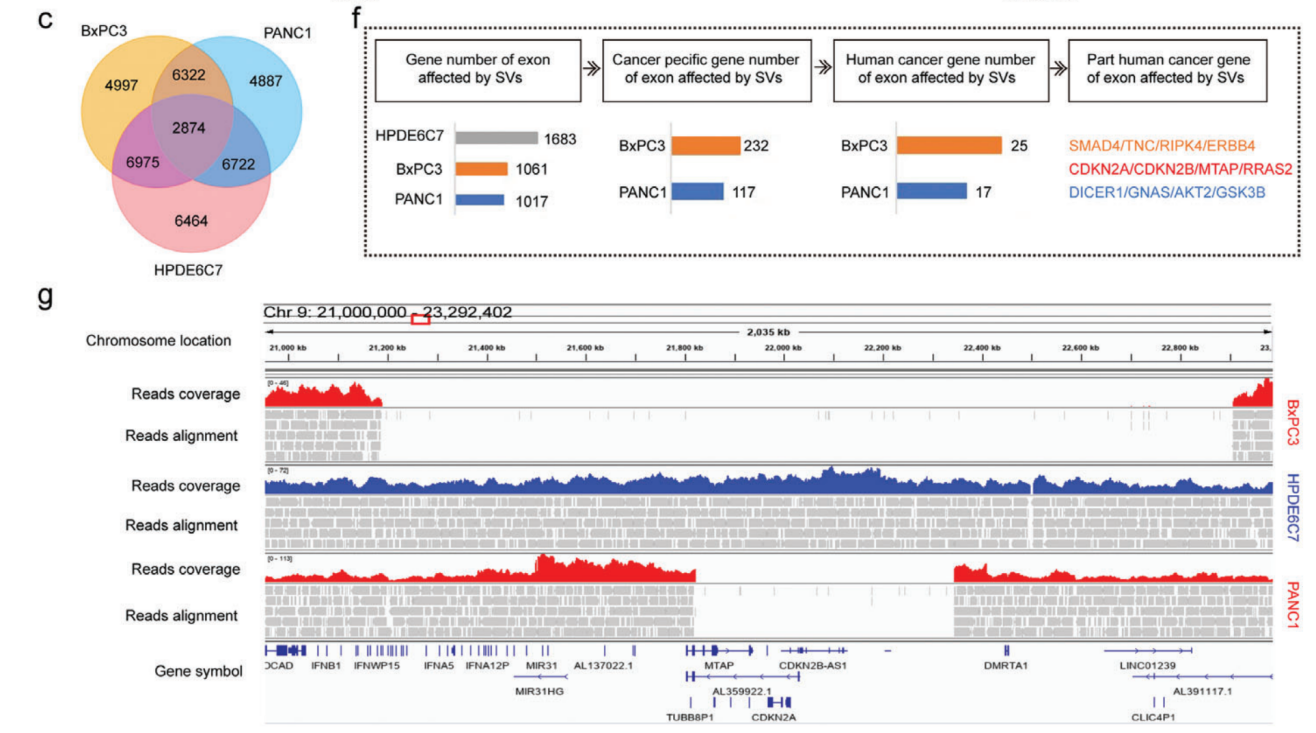
人胰腺導管癌SV影響外顯子區域基因的變化
2.染色質三維結構的廣泛重塑與人類PDAC的基因表達變化相關
Hi-C測序全面分析正常HPDE6C7細胞系和兩種癌細胞系(BxPC3和PANC1)染色體的空間構象,對來自不同庫的三個細胞系的主要讀數的相關性分析表明,來自不同庫的Hi-C數據是一致的,兩種癌細胞類型最相似,可以與正常的HPDE6C7細胞系區分開來。
PADC中染色質A/B室置換分析,與正常細胞相比,BxPC3中A/B置換的總發生率分別為24.8%(A到B,15.3%;B到A,9.5%)和PANC1中的24.1%(A到B,16.2%;B到A,7.9%)。A/B置換與基因密度和調節活性的變化有關,穩定的A和B室到A室置換是具有活性轉錄的基因豐富的區域,而穩定的B室和A室到B室置換具有相反的特性,RNA-seq數據相結合也證實這一結論。其中癌癥樣本中PHLDA1是位于12號染色體共同B到A室置換的基因,在兩個癌細胞系中都顯著上調,PHLDA1在幾種惡性腫瘤中顯著上調,包括胰腺癌、低級膠質瘤和黑色素瘤,它與胰腺癌預后不佳顯著相關。這些結果表明兩個癌細胞系(BxPC3和PANC1)中染色質A和B室的空間分布發生了變化,這些轉變與癌癥相關基因的表達變化顯著相關。
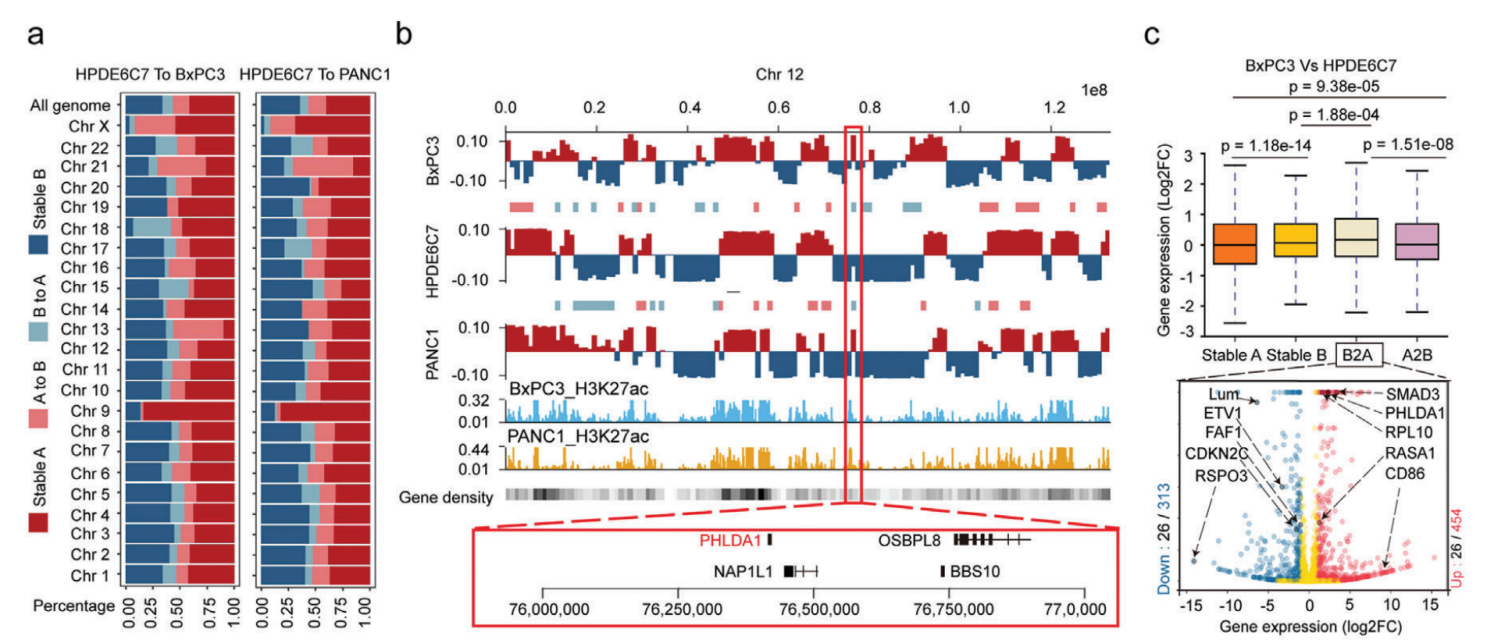
人胰腺癌染色質A/B室置換對基因表達的影響
PDAC中的接觸域(CD)改變分析,拓撲關聯域(TAD)是染色質結構的功能單位,這里利用高分辨率Hi-C(5kb)測序檢測正常細胞系和癌細胞系的接觸域邊界(CDB),在HPDE6C7、BxPC3和PANC1細胞系中確定了14 581、14 318和13 494 CD,平均大小分別為211、227和214kb。CDB保存在人類基因組中任何兩種細胞類型之間共享的CDB很少,CD數量和大小的變化可能會在不同的染色體區域出現截然相反的變化。與共享的的CD相比,癌癥特異性CD區域與上調基因表達的關聯要大得多。這些癌癥特異性CD中差異表達基因的GO富集分析顯示,改變的基因涉及幾種關鍵途徑,包括癌癥促進、細胞分化、細胞粘附、細胞運動和遷移。
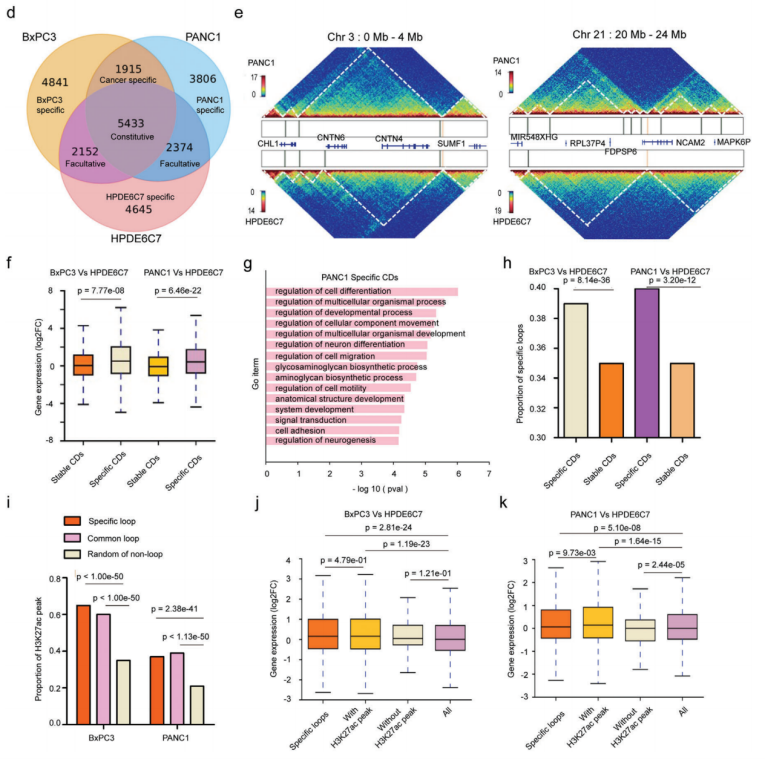
人胰腺癌染色質CDB和loops環對基因表達的影響
PDAC中的癌癥特異性loops和異常增強子激活深入研究,顯示癌癥特異性loops異常增多,癌癥特異性CD與高比例的癌癥特異性loops顯著相關,這些H3K27ac活性的癌癥特異性loops與上調的基因表達顯著相關。這些變化伴隨著基因表達的上調,可能與癌癥特異性loops中調節元素活性的增強有關。總之,PDAC細胞系中的3D染色質結構經歷了廣泛的重塑,并隨之而來的基因表達失調,這可能會促進PDAC的腫瘤發生和進展。
3、三維基因組結構變異的分布
人體內SV的發生和形成往往受到染色體三維空間結構的影響,根據SV和染色質A/B室之間的關系,除了BxPC3中的缺失外,BxPC3/PANC1/HPDE6C7樣本中染色質A室都有顯著的插入、缺失和重復變化,發現插入、其中在PANC1的染色質A室中顯著富集了缺失和重復變異,但在BxPC3的染色質A室中顯著存在重復變異,兩種癌細胞系之間A/B隔間中癌癥特異性SV的分布模式大不相同。
SV在CD中的分布及其邊界研究,根據SV和CDB斷點之間的相對位置,將所有癌癥特異性SV分為四類:CDB中/交叉CDB/內部CDB/部分重疊。超過90%的癌癥特異性SV位于CD(CDB-SV)內部,這對它們的染色質空間構象影響很小,而跨CDB/內部CDB/部分重疊SV對CD折疊的影響更大,其比例相對較低。總之,在A/B室或CD級別的腫瘤中,SVs的發生與染色體三維空間構象之間存在一定的相關性。此外,SV在三維基因組中的分布模式具有高度細胞類型特異性。
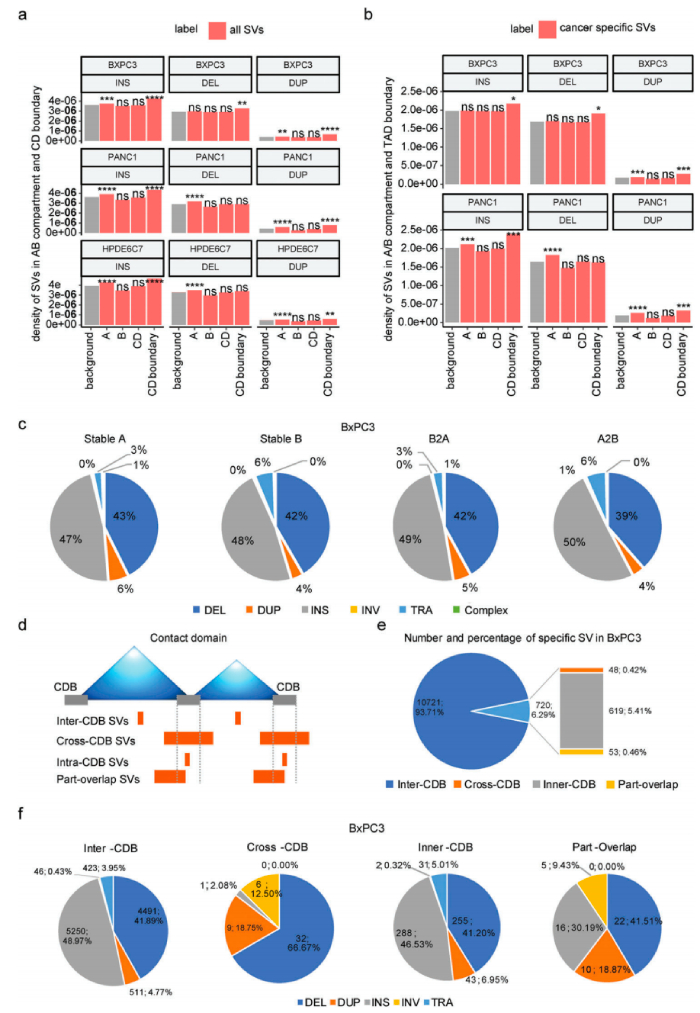
三維基因組染色質空間構象與結構變異的分布
4、PDAC基因組中特定癌癥SV和染色質結構域的相互作用
SV可以重塑線性基因組染色質空間構象,從而改變順式中的染色質拓撲和基因調控,深入探索跨CDB SV對CD中斷的影響,跨CDB缺失區域的CD融合明顯高于基因組的其他位點,并且只有同一細胞系中頻率更高的缺失與相鄰CD或CD融合的增強相互作用有關,低頻率的缺失對相鄰CD的相互作用沒有顯著影響,在這些情況下沒有發現CD融合。這些表明SV對3D基因組結構的影響相當復雜,可能會受到多種因素的影響,例如SV的細胞間基因組異質性、位置和長度等。
分析了兩個癌細胞系中CD融合與差異基因表達之間的相關性。結果表明,融合CD中差異表達基因的比例明顯高于基因組其他區域,總的來說,這些數據表明,癌癥特異性SV可以通過重塑PDAC中的CD來調節基因表達。
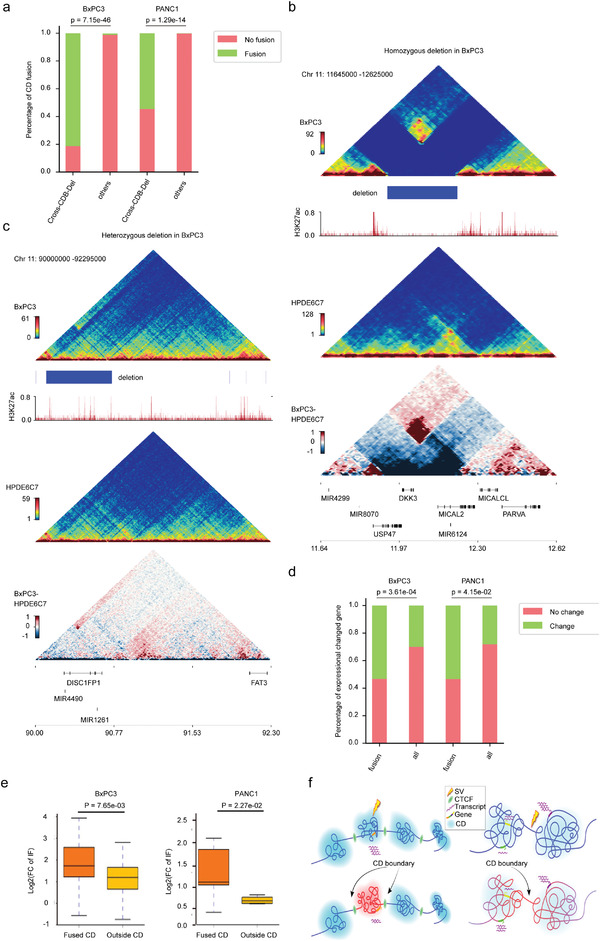
癌癥特異性SV通過重塑PDAC基因組中的CD來影響基因調控
5、CDKN2A純合子缺失對三維基因組結構和基因表達的影響
CDKN2A失活通過各種機制發生在約90%的PDAC中,其中純合子缺失是常見的途徑之一。此次研究證實了BxPC3和PANC1中CDKN2A的純合子缺失,發現缺失兩側相鄰CD之間的相互作用顯著增強,形成融合CD,相鄰CD區域之間的內部相互作用也顯著增強。發現CDKN2A、CDKN2B和MTAP(缺失區域的基因)的表達幾乎消失了,而MIR31HG和LINC01239(缺失區域兩側的基因)的表達被顯著上調。發現MIR31HG表達高患者的生存率顯著縮短(圖5e),與MIR31HG在PDAC中的致癌作用一致。該研究揭示了CDKN2A純合子缺失對三維基因組結構和基因表達的影響,為了解CDKN2A失活以推動PDAC的發生和發展提供了新的見解。
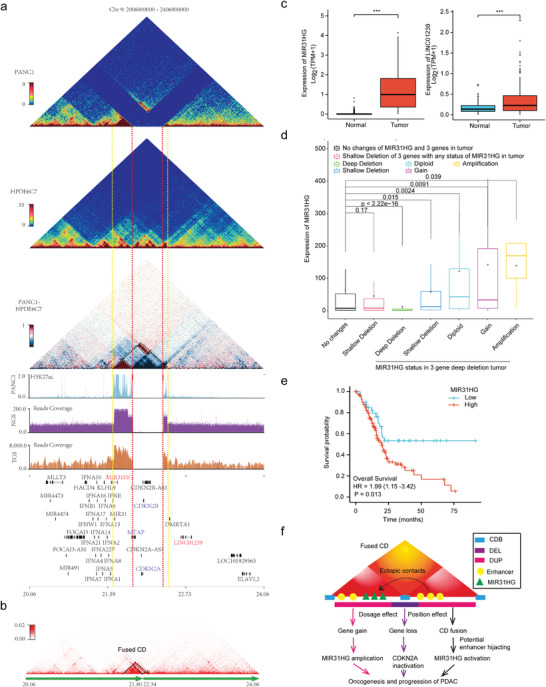
CDKN2A純合子缺失部分通過伴隨的相鄰基因組擴增和CD融合與MIR31HG上調有關
6、PDAC驅動基因SMAD4缺失對三維基因組和基因表達的影響
SMAD4是PDAC的一個關鍵驅動基因,已知在約55%的胰腺癌中丟失,純合子缺失占這些病例的約30%。通過三代測序技術和Hi-C方法驗證了BxPC3中的SMAD4純合子缺失。涉及SMAD4基因的交叉CDB缺失雙方之間的相互作用沒有增強,分析BxPC3中第18號染色體的相互作用熱圖,并發現了幾種增強的遠端異位相互作用。這些不尋常的遠程區域主要與三個大型缺失點有關,包括SMAD4缺失區域。BxPC3中SMAD4的純合子丟失和18號染色體上的多次缺失會導致巨大、復雜的染色體重排,包括反轉和易位。CD可以減少復雜染色體重排造成的3D基因組組織的破壞,保持染色質的基本結構,并穩定CD中基因的表達。確定了與18號染色體上SMAD4缺失相關的復雜染色體重排,并揭示了它們對3D基因組組織和相關基因表達的影響。

PDAC中SMAD4缺失相關的復雜基因組重排與三維基因組變化
總結
三代測序和高通量染色質構象捕獲技術(Hi-C)的快速發展和應用,越來越多的證據表明,SV和3D基因組在腫瘤發生和發育中起著關鍵作用。建立SV和3D基因組結構的特征,并表征它們在PDAC中的動態相互作用,闡明了兩個關鍵驅動基因CDKN2A和SMAD4的純合子缺失對3D染色質折疊結構域的影響,以及相關基因在PDAC的致癌和進展中的表達,這項研究為全面理解SV在胰腺癌發生和發展中的功能和致病機制提供了新的空間視角。基于高維基因組學的研究將有助于識別PDAC新的分子標記物或潛在靶點,這對提高預后極差的PDAC的治療挑戰具有重要的現實意義。
參考文獻
Du Y, Gu Z, Li Z, et al. Dynamic Interplay between Structural Variations and 3D Genome Organization in Pancreatic Cancer. Adv Sci (Weinh). 2022 Jun;9(18):e2200818. doi: 10.1002/advs.202200818.
如果您對三維基因研究感興趣,歡迎點擊下方按鈕聯系我們,我們將免費為您設計文章思路方案。
百邁客轉錄調控多組學研究一站式服務
百邁客轉錄調控事業部搭載二代Illumina Novaseq、三代Pacbio/Nanopore測序平臺的測序數據開發基因及轉錄本水平表達及功能研究的系統化解決方案。Hi-C實驗、信息分析平臺具有雙認證,為Cell、Nature genetics、National Science Review、Molecular plant等20余篇IF≥10文章提供HIC技術服務。具有50余篇基因組研究共同作者項目案例,以及不少于5篇通訊作者項目案例。與甘肅省人民醫院、四川大學華西醫院、中國醫科大學等單位合作發表文章影響因子超1500+。百邁客可以提供從研究方案制定、建庫測序、生物信息分析、定制化分析、個性化交互分析、生信培訓班的轉錄調控多維解決方案。
]]>
發表時間:2022.10.8
發表期刊:Lipids in Health and Disease
研究背景
在全球范圍內,肥胖是一個日益嚴重的健康問題。其發病率逐年增加。肥胖會導致許多并發癥,包括心血管疾病、中風、肝硬化和糖尿病。因此,新的預防性治療策略對于降低肥胖發病率至關重要。肥胖的原因包括多種原因,包括環境和遺傳因素。然而,個體似乎對肥胖表現出不同程度的易感性。高脂飲食有助于 C57BL/6 模型小鼠的肥胖或肥胖抵抗。盲腸或新鮮糞便中的微生物群和血液或尿液中的代謝物有助于肥胖抵抗;然而,小腸中的微生物群或代謝物尚未得到廣泛研究。因此,該研究旨在調查小腸中肥胖/肥胖抵抗的不同潛在新型生物標志物。
技術路線
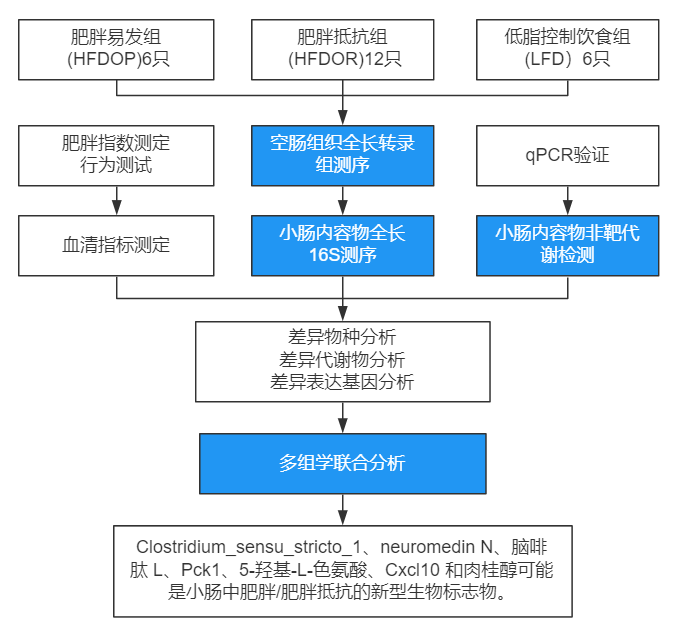
主要結果
1.肥胖/肥胖抵抗的主要指標
高脂飼料喂養8周后,單籠飼養4周,高脂飲食誘發的肥胖組(HFDOP)小鼠體重和Lee‘s指數持續增加,而肥胖抵抗組(HFDOR)小鼠體重和Lee’s指數持續增加。然而,HFDOP和HFDOR小鼠的平均每日飼料和能量攝入量沒有顯著差異(圖1B、C)。高脂飲食組與高脂飲食組小鼠體重和Lee‘s指數有統計學差異。HFDOP組的空腹血糖水平高于HFDOR組和LFD組(圖1G)。HFDOP組的肝臟重量、總脂肪和總內臟脂肪的重量顯著高于LFD組(圖1H-J).
然而,HFDOR組的肝臟、總脂肪和總內臟脂肪的重量顯著低于HFDOP組(圖1H-J)。HFDOP組AST、ALT、HDL、LDL、TG和TC水平較LFD組顯著升高(圖3K-P)。然而,HFDOR組的AST、ALT、高密度脂蛋白、低密度脂蛋白和甘油三酯水平顯著低于HFDOP組(圖3K-O)。盡管HFDOR組的TC水平低于HFDOP組,但差異無統計學意義。
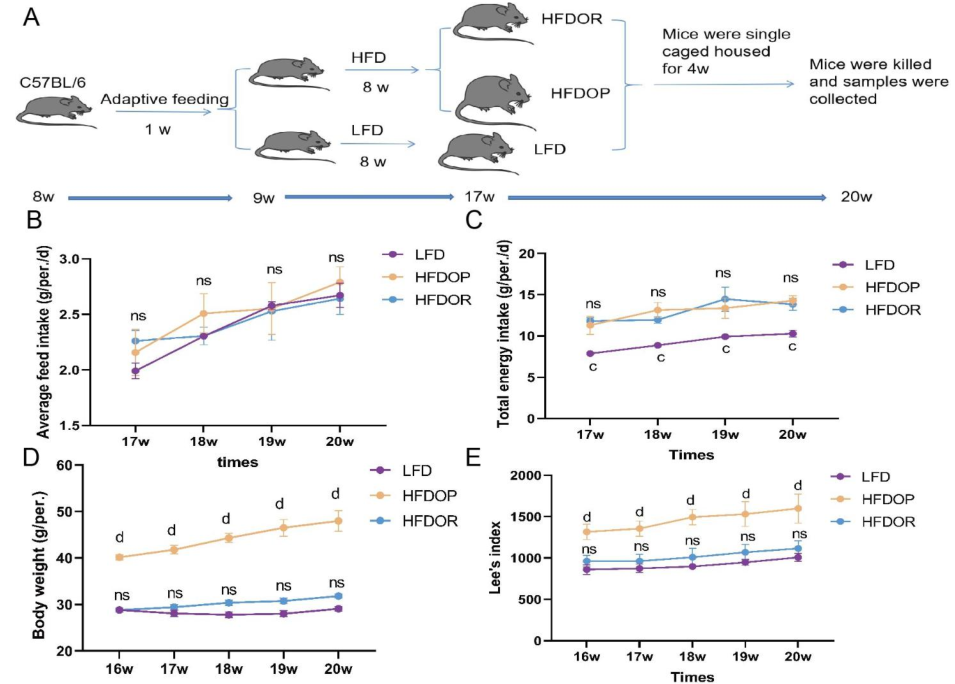
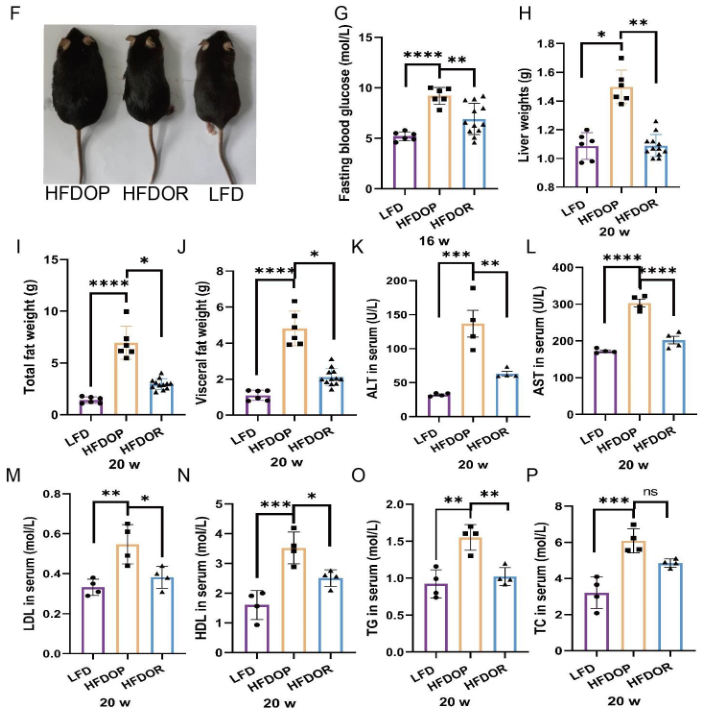
圖1.肥胖/肥胖抵抗小鼠的主要指標
2.肥胖抵抗與抑郁無關
為了排除抑郁和焦慮樣行為對肥胖抵抗的影響,在16周結束時進行了自發開場活動實驗和高架十字迷宮實驗。開場實驗結果如圖2A-C所示。HFDOP組進入中心區域的次數和進入該區域的次數顯著低于LFD組(圖2B、C)。HFDOR 組比 HFDOP 組更遠地進入中心區域并且更頻繁地進入該區域(圖2B、C)。高架十字迷宮實驗表明,與LFD組相比,HFDOP組進入開臂的次數和頻率都要少得多(圖3E、F)。HFDOR組比HFDOP組開臂走得更遠,但兩組之間沒有統計學意義。HFDOR組比HFDOP組更頻繁地進入開臂(圖3F)。
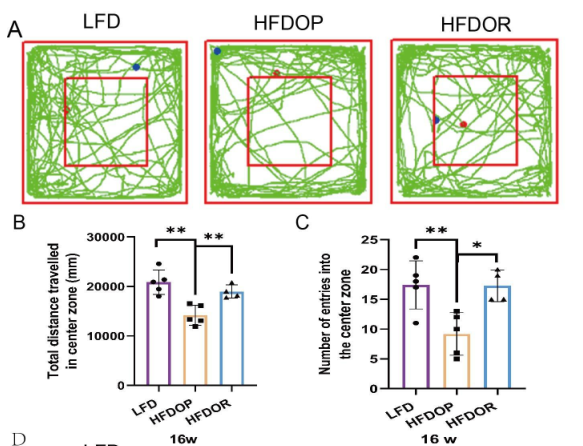
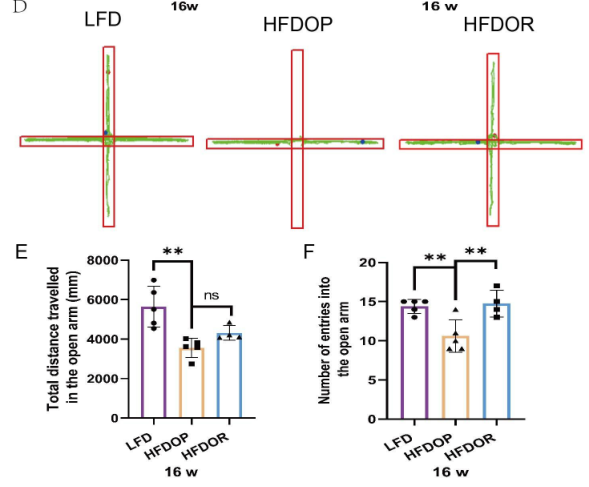
圖2.肥胖/肥胖抵抗小鼠對抑郁和焦慮樣行為測試
3.肥胖/肥胖抵抗與小腸菌群和代謝產物有關
使用全長16S微生物多樣性測序和非靶向代謝組學方法檢測了小腸內容物。HFDOP組的Muribaculaceae、Faecalibaculum、Desulfovibrio和 Lachnospiraceae的相對豐度高于 LFD 和 HFDOR 組,但差異無統計學意義。LFD對照組和HFDOR組Clostridium和Lactobacillus的相對豐度高于 HFDOP 組,但只有梭狀芽胞桿菌水平的差異具有統計學意義。
小腸內容物非靶向代謝組學的結果如圖3C-E 所示,PLS-DA 分析表明 HFDOR 和 HFDOP 組之間的明顯分離(圖3C)。在小腸內容物中共檢測到 314 種差異代謝物(圖3D)。相關性分析表明,Desulfovibrio、Bifidobacterium和Gemella都與 PC、DG 和膽酸葡糖苷酸呈正相關,而與維生素 A、dCMP、肉桂醇、5-HT 和肉桂酸葡糖苷酸呈負相關(圖3E)。Clostridium_sensu_stricto_1 和 Ralstonia 與維生素 A、dCMP、肉桂醇、5-HT、1 H-吲哚-3-乙酰胺和氫化肉桂酸呈正相關,與 PC、PE、TG、DG、neuromedin N、腦啡肽 L 和膽固醇葡糖苷酸呈負相關(圖 4)。

圖3.小腸菌群和代謝物中肥胖/肥胖抵抗的差異生物標志物
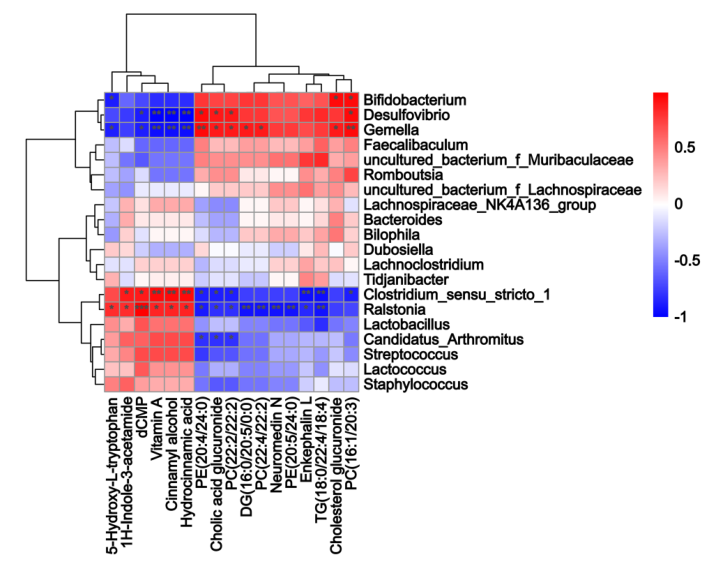
圖4. 小腸微生物群與代謝物的 Spearman 相關性分析
4.肥胖/肥胖抵抗力的小鼠在空腸中表現出不同的轉錄水平
為了進一步探討肥胖/肥胖抵抗的原因,研究者運用全長轉錄組測序技術檢測空腸組織中的基因轉錄水平。共鑒定出1645個差異表達基因(750個上調,895個下調),其中331個差異表達顯著。在331個顯著差異表達基因中中,有30個基因在11條KEGG通路中被富集。富集的基因和10條最豐富的KEGG途徑如圖所示,這些基因在絲裂原活化蛋白激酶(MAPK)、細胞因子和磷脂酰肌醇3’激酶/蛋白激酶B(PI3K/Akt)信號通路中顯著豐富(圖6C)。用qPCR分別驗證了差異表達基因。Tgfb2、Spp1、Pck 1、Cxcl10和Epha7表現出一致的mRNA表達模式(圖6B,D-H)。
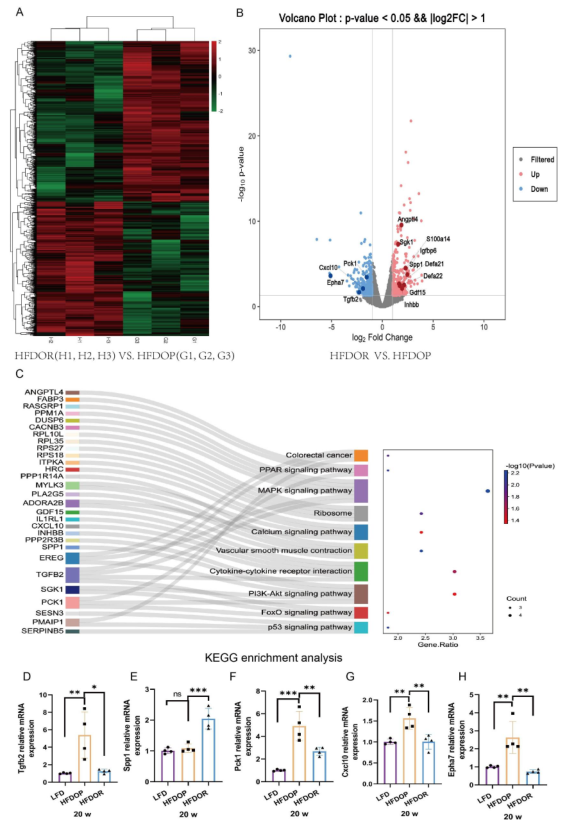
圖6.空腸中肥胖/肥胖抵抗的不同轉錄水平
5.差異表達基因與代謝物或微生物群的相關性分析
進一步進行了DEGs和微生物的Spearman相關性分析。結果表明,esulfovibrio, Bifidobacterium和Gemella與CxCl10呈正相關,與SGK1、ANGPTL4和GDF15呈負相關。
Clostridium_sensu_stricto_1 和 Ralstonia 與 Igfbp6 和 Gdf15 呈正相關,與 Epha7、Pck1和Cxcl10 呈負相關。此外,Defa21和Defa22與uncultured_bacterium_f_Muribaculaceae呈負相關(圖7A)。DEGs與代謝物的相關分析結果表明,H-吲哚-3-乙酰胺與Igfbp6和Inhbb水平呈顯著正相關,與Epha7呈負相關。代謝產物5-羥色胺與SGK1呈顯著正相關。維生素A、肉桂醇和氫肉桂酸與GDF15呈顯著正相關,與CxCl10呈負相關。代謝產物PC、PE、TG、DG、腦啡肽L、NeuroMedin N、膽酸葡萄糖醛酸苷和膽固醇葡萄糖醛酸苷與CxCl10、Pck 1、Epha7和Tgfb2呈顯著正相關,與Inhbb、Igfbp6、S100a14、GDF15、ANGPTL4和SGK1呈負相關(圖7B)。
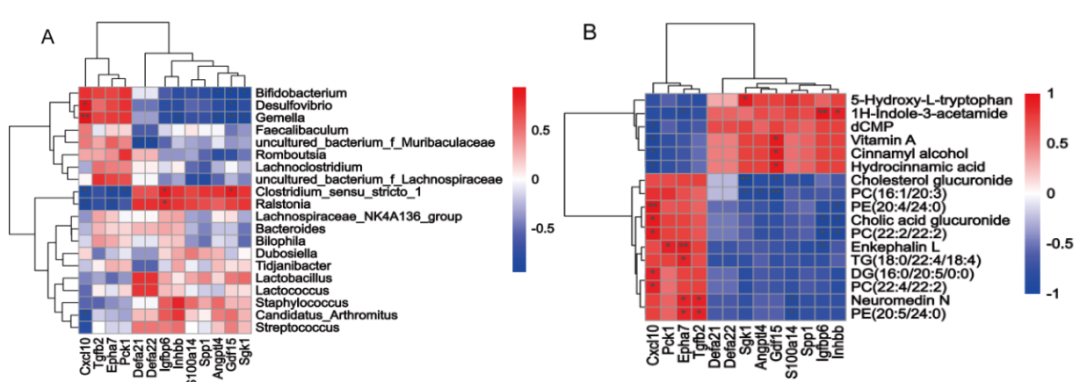
圖7.差異表達基因與腸道菌群和代謝物的 Spearman 相關性分析 A.差異表達基因與小腸菌群相關性分析; B 差異表達基因與小腸代謝物相關性分析。
總結
綜上所述,腸道菌群、腸道代謝產物和腸道基因相互作用。腸道微生物群Clostridium、Desulfovibrio和Lachnospiraceae可直接作用于腸粘膜,其代謝產物5-羥色胺(5-HT)、腦啡肽L和神經介素N可能調節神經系統并將信號傳遞給大腦。因此,“微生物-腸道-大腦”軸可能與肥胖抵抗有關。此外,Cxcl10也是HFD誘導的ENS損傷的潛在靶點。未來預防和治療肥胖的目標可能包括5-羥色胺、腦啡肽L和神經介素N、肉桂醇和1H-吲哚-3-乙酰胺。益生菌補充劑,特別是梭狀芽孢桿菌,可能有助于治療肥胖患者或預防肥胖。
如果您對該研究思路感興趣,歡迎點擊下方按鈕聯系我們,我們將免費為您設計文章思路方案。

參考文獻
Pang Y, Zheng Y, Yang N, et al. Potential novel biomarkers in small intestine for obesity/obesity resistance revealed by multi-omics analysis[J]. Lipids in health and disease, 2022, 21(1): 1-15.
]]>
隨著高通量測序技術的發展,組學(Omics)研究不斷深入,通過對各組學進行高通量測序并對數據整合研究,可以全面和系統地了解基礎研究、分子育種、臨床診斷和藥物研發等領域中多種物質的相互關系。為網絡生物學、系統生物學的研究提供重要的技術手段。
隨著動物基因組數據的積累,研究者開始整合分析代謝組學與基因組、轉錄組、表型組、表觀組數據,嘗試構建出“遺傳標記或基因-代謝分子-表型”的關系網絡,從而篩選相關的生物標記,同時進一步解析相關性狀的遺傳機制。多組學技術應用于篩選到的基因和代謝物作為生物標志物,應該于標記輔助選擇中提供選擇的準確性。
本次我們給大家帶來利用多組學工具助力動物生長發育和遺傳育種調控機理研究的高分文章解讀,希望能給老師后續的研究提供思路。
代謝組學+微生物學
代謝組學+轉錄組學
代謝組是生物體發育和生理狀態在代謝水平的體現,是基因組與表型組之前的橋梁,差異積累的代謝物可以輔助時序表達的眾多基因進行“共表達”分析,提示基因功能,研究分子生化機制,將基因與表型聯系起來。
轉錄組+代謝組的多組學分析,可以同時實現從“因”和“果”兩個層面來探究生物學問題,可以從大量轉錄本信息中快速鑒定代謝相關的功能基因,構建核心調控網絡,找出關鍵候選基因,闡述生物學現象。
代謝組學+蛋白質組學
代謝組學目的是系統研究代謝中涉及的化學過程;蛋白質作為酶可以調控生物體代謝過程,影響生物體內代謝物濃度。通過整合分析。一方面可以結合兩個組學的分析結果,進行相互驗證,提高后續實驗驗證成功率;另一方面也有助于相互補充,并且使研究更加系統。
實現對生物變化大趨勢與方向的綜合了解,提出分子生物學變化機制模型,并篩選出重點代謝通路相關的蛋白質或者代謝產物,從而為后續進行深入實驗與分析提供數據基礎。
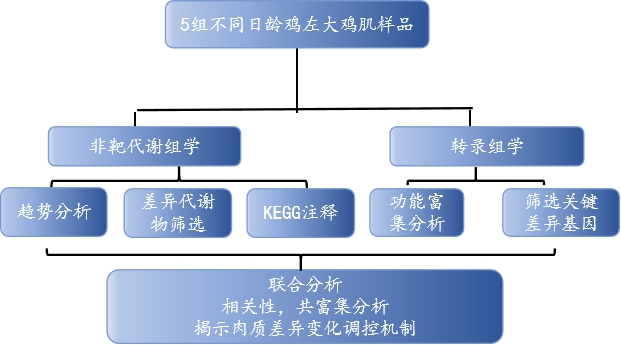
多組學關聯分析文獻案例一
Integration analysis of metabolome and transcriptome profiles revealed the?age-dependent dynamic change in chicken meat.Food Research International (IF=6.475) 2022年6月代謝組和轉錄組的整合分析揭示不同生長發育時期雞的肉質依賴性動態變化研究方案
材料:選擇20頭泌乳中期奶牛,連續8周添加20 g m/d的RPM,
方法:宏基因組測序+非靶代謝組檢測
研究背景
雞胸肉含有不飽和脂肪酸和較低的脂肪、鈉和膽固醇,是大家最喜歡肉類之一。近年來,隨著人們對食品營養價值和自身健康的重視,研究者們開始致力于如何提高肉類質量的研究,而生長時期是影響雞肉質量的重要因素,但不同時期哪些特定代謝物積累是影響雞肉品質的關鍵還不清楚。本研究使用代謝組和轉錄組技術,確定了特定代謝物積累的關鍵基因,有助于了解肉類質量發展下的生物過程,并探索特定代謝物積累的有價值的生物標志物。
技術路線
主要研究結論
隨著日齡的增長,十五烷酸、硬脂酸、肌酸、肌肽、IMP、L-組氨酸和賴亮氨酸呈上升趨勢,而鵝氨酸、DHA、天冬氨酸、LPA 18:1和 LPI 18:1隨著日齡的增長而下降。
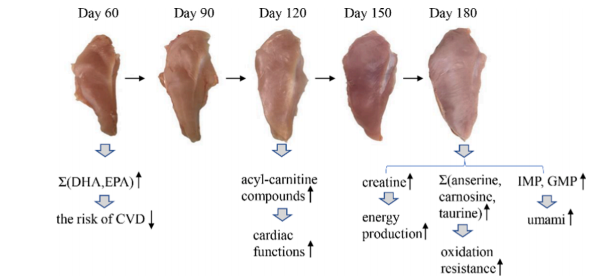
雞胸肉代謝受日齡影響的主要途徑是果糖和甘露糖代謝、花生四烯酸代謝、甾體激素生物合成、核黃素代謝、不飽和脂肪酸生物合成和亞油酸代謝。

代謝組和轉錄組的整合分析揭示了影響化學成分和代謝途徑的潛在功能基因,例如DGAT2、CYP2D6、APOV1、PLTP、PNMT。這些結果將有助于了解肉質發展的生物學過程,并探索特定代謝物積累中有價值的生物標志物。
多組學關聯分析文獻案例案例二
Interaction Between the Intestinal Microbial Community and Transcriptome?Profile in Common Carp (Cyprinus carpio L.).Frontiers in microbiology(IF 6.064),2021年4月
微生物和轉錄組聯合分析解析鯉魚腸道菌群與轉錄組水平的相互作用
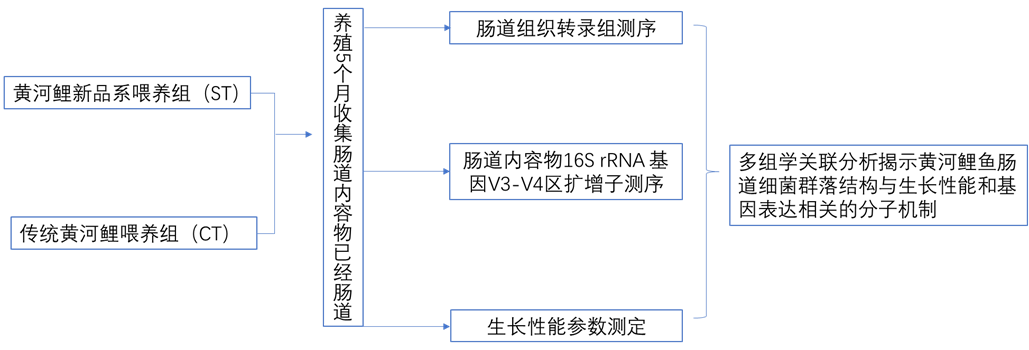
研究方案
材料:黃河鯉新品系(中國水產科學院淡水漁業研究中心)和傳統黃河鯉,水池中單獨喂養。
方法:轉錄組+16s微生物多樣性
研究背景
近年來,腸道微生物對宿主生產性能的影響受到廣泛關注。宿主的生產性能可以通過調節腸道微生物結構來調節,而腸道微生物結構又主要受遺傳和飼料的影響,腸道微生物也能影響宿主的基因表達和甲基化水平。研究發現約10%的宿主轉錄組受微生物調節,主要包括免疫、細胞增殖和代謝功能相關基因。腸道微生物與宿主基因表達之間的相互作用機制會影響飲食行為、消化過程、免疫功能和其他生理現象。一天中微生物的活動會改變宿主的生物節律、表觀遺傳學和代謝產物。當微生物群落內穩態的節律被破壞時,宿主的正常染色質和基因表達水平將發生變化,腸-肝軸基因表達的新機制將被激活,這種相互作用主要通過腦和肝的遠程控制模式實現。因此,作者運用轉錄組和微生物多樣性聯合分析來解析新黃河鯉品種的腸道微生物群通過調節腸道基因表達影響其生長性能的方式,以此為黃河鯉新品種的選育提供參考。
技術路線
不同品種鯉魚腸道轉錄組與腸道微生物多樣性研究思路導圖
主要研究結果
腸道細菌群落結構與生長性能的關系:測量了黃河鯉新品四個生長性能參數(體重、長度、寬度和深度)。對照組的所有參數均比對照組顯著提高:重量14.58%,深度7.14%,寬度5.04%,長度5.07%。為了評估黃河鯉新品種的生長性能是否與其腸道菌群有關,將實驗組和對照組在相似的生長環境中進行培養,并探討其腸道細菌群落結構的差異。多樣性分析結果顯示實驗組的平均OTU豐富度(322.80±67.87)高于對照組(303.00±53.00)。實驗組和對照組共鑒定出94個共有的細菌類群。PCA 分析和系統發育樹將所有樣本分為兩部分,表明實驗組和對照組是可明顯區分的。在屬水平上對細菌組成(相對豐度)進行分析,結果表明氣單胞菌(Aeromonas)是對照組的優勢類群,其次是厚壁菌(Firmicutes)和玫瑰單胞菌(Roseomonas),而實驗組中,玫瑰單胞菌(Roseomonas)是優勢類群,其次是厚壁菌(Firmicutes),然后是氣單胞菌(Aeromonas)。結果表明這兩個群體的細菌群落結構是不同的,這意味著宿主(黃河鯉魚新品種)基因組與微生物組相互作用,以選擇某些微生物類群,暗示腸道菌群組成會影響鯉魚的生長性能。
實驗組和對照組之間的差異基因表達:使用與16S測序相同的樣本對黃河鯉新品種(實驗組)和對照組中的轉錄組進行測序并鑒定差異基因(DEG)。在249個顯著DEGs中,194個基因表達下調,55個基因表達上調。通過GO注釋,這些基因被分為以下功能類別:生物調節、細胞過程、解毒作用、發育過程、免疫系統進程等(都屬于生物過程);細胞、細胞部分、胞外區域、胞外區域部分、大分子復合物等(細胞成分);抗氧化活性、結合活性、催化活性、分子功能調節器上等(分子功能)。聚類分析表明,實驗組和對照組具有不同的特征,DEG分為四個部分,大部分基因注釋到了免疫相關途徑。
與腸道細菌群落組成相關的差異表達基因:Pearson相關性用于推斷微生物屬級群落組成與DEGs之間的關系,共探索了2892對,包括245個基因和256個屬。篩選出來細菌群落具有屬級關系的前10個候選基因,其中許多參與免疫反應,如H-2類組織相容性抗原、類 E-Sβ鏈(H2-Eb1)、類胸苷磷酸化酶(TYMP)、干擾素誘導蛋白44(IFI44)和主要組織相容性復合物I類相關基因蛋白樣(MR1)等。DNA修復蛋白RAD51同源物4(Rad51d)、ETS易位變異體5、轉錄本變異體X2(Etv5)和著絲粒蛋白Spc24(Spc24)與細胞分化和生長性能相關,這些基因可能在黃河鯉魚新品種生長性能中發揮關鍵作用。總而言之,腸道細菌可以影響腸道中的基因表達,其中優勢種或細菌結構可能反映宿主黃鯉魚新品種的遺傳特征。腸道的細菌-基因表達譜有助于宿主腸道的健康和性能,通過與基因頻率配對確定前10個屬為:Bordetella,Lutispora,Methylocystis,Ohtaekwangia,Roseomonas,Shewanella,GpVI,Desulfovibrio,Candidatus_Berkiella and Azorhizobium,其中,Methylocystis數量最多,在甲烷循環中起作用。該研究提出了腸道細菌群落與基因表達相互作用的證據,共探索了2892對(屬級基因和細菌),包括245個基因256個屬。作者發現大多數基因涉及免疫學、細菌群落和細胞分化,其中大多數位于免疫相關信號通路中。該研究表明黃河鯉新品種生長性能的改善可能與其免疫反應的改善及其在腸道細菌結構中的相互作用有關。

組學在植物中的研究可以深入到農林畜牧,食品生產,生物醫藥,污染治理等各個方面,因此關于植物調控機制及其與環境間相互作用關系的研究,一直是組學研究的重點。
在中心法則中,RNA處于重要的中央樞紐地位,轉錄組作為生信科研中的“萬金油”,幾乎可以與所有的組學產品聯合分析。轉錄組代表了基因表達的中間狀態,可以反映諸如轉錄調控、轉錄后調控的機理;蛋白組代表生物體直接功能執行狀態,可以反應轉錄本真實的表達情況;代謝組可以反映生物體表型的狀態變化。利用多組學測序,我們可以對植物的生長發育機制、生理代謝調控、逆境脅迫響應、生物侵染反應、作物產品生產品質等方面進行深入全面的研究,精準錨定調控關鍵性狀的調控因子,為后續實際應用提供堅實基礎。
本次我們給大家帶來三篇利用多組學工具助力植物品質性狀(色澤、氣味、味道等)調控機理研究的高分文章解讀,希望能給老師后續的研究提供思路。
轉錄組+蛋白組
- 轉錄組代表了基因表達的中間狀態,可以反映諸如轉錄調控、轉錄后調控的機理。蛋白質是生物體直接的功能執行者,反映了生物體的狀況。 轉錄組學和蛋白質組學聯合分析能夠真正觀察到mRNA-蛋白質關聯性,充分利用轉錄組和蛋白質組研究的差異性和互補性,對基因的表達水平進行衡量,以獲得基因表達各個步驟表達和調控的全景圖,發掘常規單個組學未能發現的新結果。全面探究生物體疾病機理、脅迫機制,研究重要基因的表達模式和調控機理。
轉錄組+代謝
- 轉錄組的表達不能直接證明表型是否發生變化,但是表型性狀的微小變化在代謝水平會呈指數放大,可以利用代謝組來反映表型的狀態變化,但是單獨代謝組檢測,無法解釋影響表型的基因機理。轉錄組+代謝組的多組學分析,可以同時實現從“因”和“果”兩個層面來探究生物學問題,相互間進行驗證,從海量的數據中篩選出關鍵基因、代謝物及代謝通路,深度解析生物系統的宏觀發育過程,解釋生物過程的復雜性和整體性,提高文章的水平。
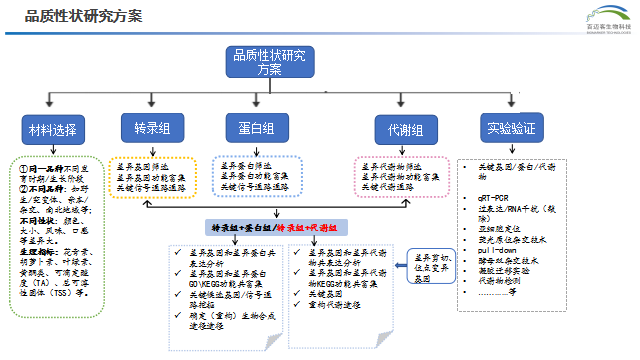
品質性狀研究方案流程圖
多組學研究在植物領域中的應用案例1、全長轉錄組和代謝組聯合分析揭示了牡丹花黃色色素沉著的調控機制
英文標題:Integrating full-length transcriptomics and metabolomics reveals the regulatory mechanisms underlying yellow pigmentation in tree peony (Paeonia suffruticosa Andr.)flowers
期刊:Horticulture Research
影響因子:7.291(2021年11月)
研究策略:全長轉錄組+靶向代謝組
Doi:10.1038/s41438-021-00666-0
實驗設計
實驗材料:黃花品種“海黃”和紫紅色花品種“肉芙蓉”牡丹花瓣。

實驗方法:分別選取五個不同開花階段(S1:第1階段,無色緊芽;S2:第2階段,輕微著色的軟芽;S3:第3階段,最初開花;S4:第4階段,半開花;S5:第5階段,花藥外露,完全開放并著色。);驗證實驗:qRT-PCR 、亞細胞定位、過表達、雙熒光素酶系統。
測序策略:全長轉錄組測序(PB平臺)+ 二代轉錄組測序 + 黃酮靶向代謝組學
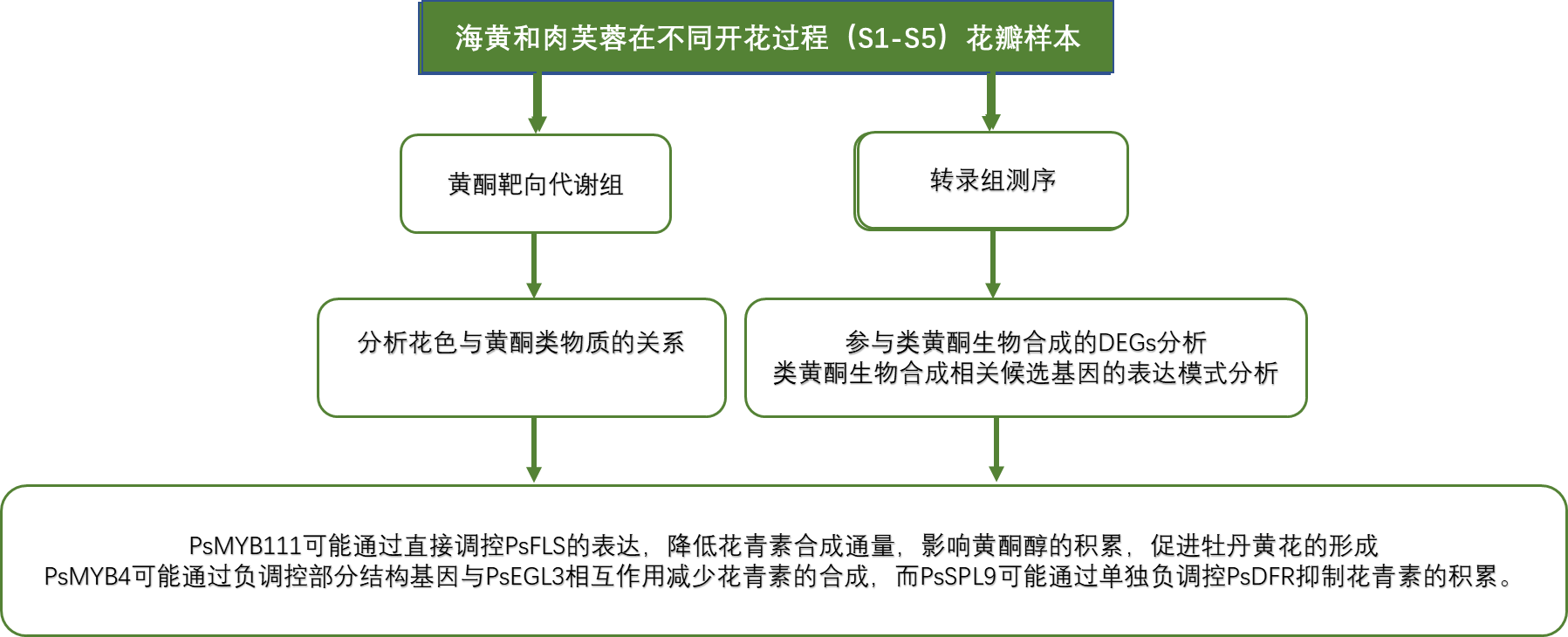
技術路線
研究內容
為表征樹牡丹花色發育情況,以黃花品種“海黃”和紫紅花品種“肉芙蓉”為研究對象,在花蕾早期至盛開的S1-S5共5個發育階段對兩個品種的花瓣組織進行了取樣,用于測定花瓣顏色指數,在“海黃”中發現L *(明度)值從S1到S5逐漸增加,這表示花瓣顏色明度升高。C *(色度)和b *(黃色)在S3達到峰值,隨后在S4和S5下降,證明S3是顏色最黃的階段。而“肉芙蓉”在整個開花過程中沒有觀察到L *的顯著變化。在S1到S5階段,“肉芙蓉”中b *顯著低于“海黃”。同樣,“肉芙蓉”中a *(紅色)比“海黃”要高得多。
之后作者采用靶向代謝組技術測定了花瓣中的黃酮含量,“海黃”中靶向黃酮類化合物在S1至S3階段顯著增加,然后在S4和S5略微下降。在S3-S5開花后期階段,THC的含量顯著高于其他黃酮類化合物。Ap和Km在五個開花階段都有相似的黃酮含量。在整個開花過程中,“海黃”中沒有檢測到花青素。相比之下,在“肉芙蓉”中檢測到了三種花青素,其中主要是芍藥素3-O-葡糖苷(Pn3G),在S5階段達到高水平,說明Pn3G可能有助于紫紅色著色。與“海黃”相比,THC、Is、Ap含量在前4個階段快速上升,在S5階段下降,變化范圍比花葵素3-O-葡糖苷(Pg3G)的變化范圍相對較小。相反,Lu、Km、Qu、花青素3-O-葡糖苷(Cy3G)的含量沒有明顯的變化,推測這些組分可能對于紫紅色著色沒有顯著影響。
全長轉錄組分析結果顯示,“海黃”黃酮類生物合成基因在S2階段比“肉芙蓉”表達水平高,分別為PsCHSs和PsCHIs,PsCHS在合成THCs中有重要作用,而PsCHI在黃酮合成過程中發揮關鍵作用,這有助于黃色著色,此外,注釋為PsF3Hs的基因也有很高的轉錄水平。在這些基因中,PsFLS可以改變黃酮類途徑,以促進黃酮醇的合成。“海黃”和“肉芙蓉”中這些表達的變化與“海黃”中高水平的THC、黃酮和黃酮醇一致。S2 vs?S3階段,發現“海黃”中PsDFRs和PsUFGTs表達水平顯著下調,PsDFR和PsUFGT在花青素合成中起關鍵作用,如Cy3G,Pn3G和Pg3G的生物合成。因此,PsDFRs和PsUFGTs在“海黃”表達下調可以解釋這種黃色花卉品種中缺乏花青素的產生。
TF分析顯示調節黃酮類生物合成的TF基因都在具有黃色花的“海黃”品種中上調,MYB和bHLH是調節植物黃酮類生物合成的關鍵轉錄因子TFs。該研究鑒定了幾個差異表達的MYB和bHLH轉錄因子,每一個都與牡丹黃酮類的產生有關。其中,PsMYB4被鑒定為具有bHLH相互作用位點的負黃酮類調節劑,這意味著它們可能形成一個復雜的負調控網絡。研究發現PsMYB111通過單獨調節PsFLS促進黃酮醇的積累。此外,PsMYB4和PsEGL3可能形成復合物負調控部分結構基因,而PsSPL9可能單獨負調控PsDFR,抑制花青素的產生,降低藍色著色。
亞細胞定位分析與轉基因實驗結果進一步驗證了上述基因的關鍵作用:亞細胞定位分析顯示PsMYB111定位于細胞核,表明PsMYB111可能在細胞核中發揮TF的作用。PsMYB111在煙草中的過表達使其花顏色由玫瑰紅變為淺粉色。轉基因煙草株系中Km、Qu等黃酮醇含量顯著增加,而Pg3G、Pn3G等花青素含量顯著降低,證實了其在黃色牡丹花著色中的作用。此外,在GhMYB1a過表達的轉基因煙草株系中,NtCHS、NtF3H和NtFLS的表達顯著上調,GhMYB1a顯著激活了非洲菊GhDFR和GhMYB10的NtCHS和NtFLS啟動子。同樣,PsMYB111對PsCHS和PsFLS啟動子,尤其是PsFLS有顯著的激活作用。

牡丹黃色素沉淀調控機制
多組學研究在植物領域中的應用案例2、多組學聯合解析突尼斯軟籽石榴揮發性風味物質生物合成機制
英文標題:Transcription profile analysis for biosynthesis of flavor volatiles of Tunisian soft-seed pomegranate arils ??????
期刊(IF):Food Research International
影響因子:7.425(2022年4月)
研究策略:轉錄組學+非靶代謝組(GC-MS)+生理指標測定
Doi:10.1016/j.foodres.2022.111304
實驗設計
實驗材料:分別采集來自云南大理市(Y_DTN)、云南麗江市(Y_LTN)、云南建水縣(Y_JTN)、云南曲靖市(Y_QTN)、四川會理市(S_DHT)和攀枝花市(S_PTN)以及河南滎陽市(H_HYT)共7個不同產區的石榴。
實驗方法:取果肉進行轉錄組和代謝組測序。生理指標測定:pH值、有機酸、可溶性糖(TSS)、維生素C含量
測序策略:轉錄組(Illumina測序平臺)+非靶代謝組(GC-MS)
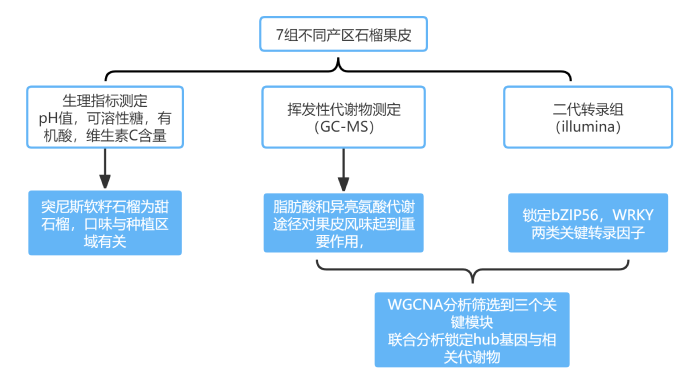
技術路線
研究內容
風味是影響石榴及其產品感官的重要因素,在石榴的加工和貯藏過程中很容易失去新鮮度,但目前對風味相關化合物的生物合成還知之甚少。
該研究對中國7個地區突尼斯石榴的代謝組和轉錄組、風味相關屬性和揮發性化合物進行了研究。7個不同種植區的石榴,糖、有機酸和維生素c含量存在顯著差異。所有樣品共鑒定了40種揮發性化合物,其中13種揮發性化合物。所有樣品中有4種化合物含量較高,包括1-己醇、(Z)-3-己烯醇、α-松果醇和2,4-二叔丁基苯酚,是影響石榴香氣品質的主要貢獻因素。所有樣品的5個差異積累代謝物,包括棕櫚酸、辛酸、月桂酸、癸酸和肉豆蔻酸,在所有樣品的脂肪酸生物合成中均顯著富集。WGCNA分析結果表明42個候選風味相關差異表達基因和9個轉錄因子主要位于3個關鍵模塊中。己烷酸是一種重要的代謝物,與38個差異表達基因顯著相關。本研究構建了復雜的調控網絡,以確定調控石榴中揮發性化合物代謝的結構基因和轉錄因子,發現bZIP56、bZIP56、bZIP20、WRKY24和bHLH9在石榴果皮的風味代謝調控中起重要作用。
該研究為理解石榴果皮風味生物合成和調控網絡的差異提供了新的見解。對于突尼斯軟籽石榴,生長區環境因素對不同生長階段風味品質調控的干預機制有待進一步探討。
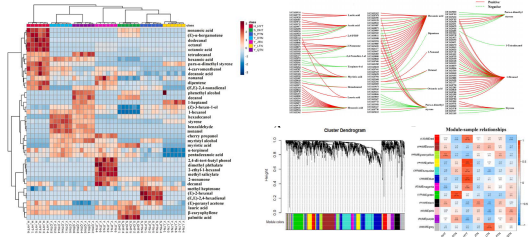
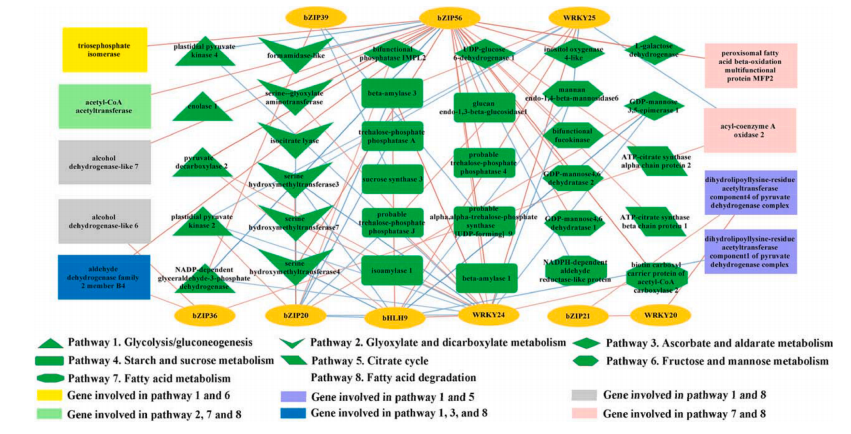
代謝+轉錄組解析石榴果風味調控機制
多組學研究在植物領域中的應用案例3、代謝組+轉錄組聯合分析揭示了紫茶花中苯類-苯丙烷色素和香氣的關系
英文標題:Integration of Metabolome and Transcriptome Reveals the Relationship of Benzenoid-Phenylpropanoid Pigment and Aroma in Purple Tea Flowers
期刊(IF):Frontiers in plant science
影響因子:5.753(2021年11月)
Doi:10.3389/fpls.2021.762330
實驗設計
實驗材料:在廣東省白塘鎮的茶園中培育了紫茶花BT和白花茶花ZJ
實驗方法:采摘第2階段(開花前)的花朵,收集花瓣液氮速凍,-80℃保存。
測序策略:轉錄組(Illumina測序平臺)+非靶代謝組(GC-MS)
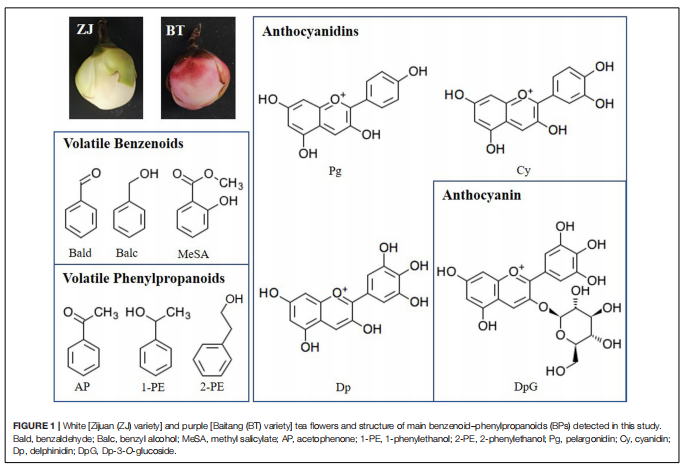
主要研究內容
山茶花的花通常是白色的,葉片是紫色的,研究發現了一種特殊的品種,葉子和花都是紫色的,而研究者通常關注顏色形成的機制,而忽略了香氣的變化。紫茶樹含有更多的花青素,屬于黃酮類化合物。同時,由莽草酸途徑衍生的苯丙氨酸(Phe)是黃酮類化合物和揮發性苯類苯丙素(BPs)的前體。因此,目前尚不清楚BP的香氣是否因紫色的出現而減弱。
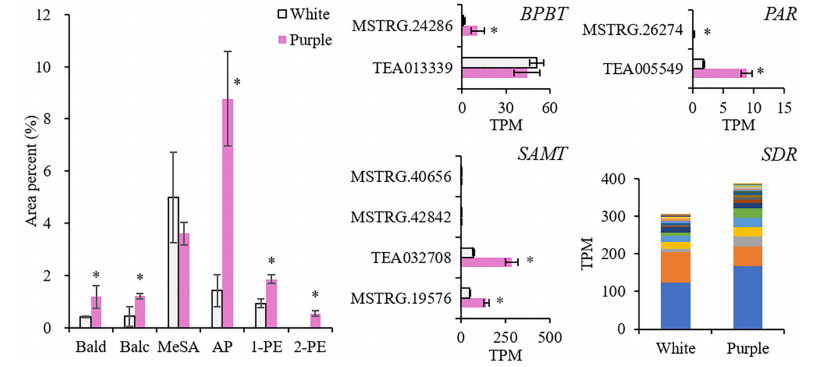
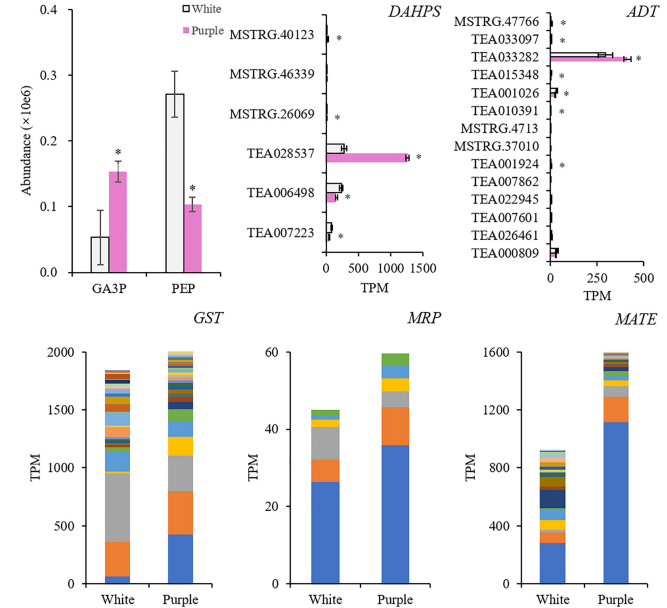
本研究整合了白花(ZJ)和紫花(BT)的花瓣代謝組和轉錄組,揭示了顏色(花青素)和香氣(揮發性BPs)之間的關系。結果表明,在紫色花瓣中,3-脫氧-d-阿拉伯七甲糖酸7-磷酸合酶(DAHPS)促進的上游莽草酸途徑升高。在增加的花青素中,飛燕草素-3-O-葡萄糖苷(DpG)極高;苯甲醛、苯乙醇、苯醇(AP)也增強,AP顯著升高。誘導揮發性BPs生物合成相關的結構基因,在整個類黃酮生物合成途徑下調,不同的是,類黃酮F3’?H和類黃酮F3’?5’?H通過高表達,將碳通量轉移到飛燕草素,然后通過增加青銅-1(BZ1)(UDP-葡萄糖:類黃酮3-O-葡萄糖基轉移酶)與糖苷結合形成DpG。
選擇與AP和DpG高度相關的轉錄因子(TFs),研究它們與差異表達的結構基因的相關性。結果顯示MYB、AP2/ERF、bZIP、TCP、GATA等基因顯著表達,并專注于Phe上游合成基因的調控(DAHPS;和AP(苯乙醛還原酶、短鏈脫氫酶/還原酶)、Dp(F3’H;F3’?5’?H)和DpG(BZ1)的合成,但抑制黃酮(黃酮醇合酶)和兒茶素(白花青素還原酶)的形成。這些結果發現了紫茶樹中揮發性BPs的促進作用,擴展了我們對BPs型顏色與香氣關系的理解。
如果您對以上測序思路感興趣,歡迎點擊下方按鈕聯系我們,我們將免費為您設計文章思路方案