研究背景
胎生是脊椎動物中廣泛存在的一種生殖策略,其特征為受精卵在母體內發育,直至胚胎發育成熟后才被產出。與這一普遍模式形成鮮明對比的是,海馬科物種演化出了極為特殊的“雄性懷孕”現象,該獨特的生命史策略為研究動物生殖系統的演化提供了重要窗口:功能性相似的生殖方式究竟源于趨同的分子機制,抑或是通過同源通路或全新的細胞調控路徑實現?盡管已有比較基因組學研究初步揭示了該生殖方式轉變的遺傳背景,然而相關基因在特定細胞類型中的表達模式及其演化軌跡仍不清楚。
在結構上,海馬育兒袋與有袋類動物的育兒袋具有一定相似性,并在功能上融合了羊膜動物子宮與胎盤的特點。在雄性懷孕過程中,其育兒袋內層組織發生顯著肥大與血管化,形成一種被稱為“偽胎盤”的結構,可執行氣體交換與營養物質傳遞等典型的胎盤功能。此外,海馬在進化過程中丟失了?foxp3?基因——該基因在哺乳動物中對調節性T細胞(Treg)的發育與功能維持具有核心作用,這一遺傳缺失也引發了對其在懷孕過程中免疫耐受機制的特殊適應性的探討。
研究內容和結果
對海馬育兒袋7個發育階段進行單細胞轉錄組測序,鑒定出14個細胞簇,分為四大主要細胞類型:上皮細胞(EPCs,9個簇)、成纖維細胞(FBs,2個簇)、免疫細胞(ICs,2個簇)和內皮細胞(ENCs,1個簇)。細胞的空間分布對細胞間相互作用及其功能維持至關重要。
對妊娠早期胎盤囊進行空間轉錄組測序(測序平臺BMKMANU?S1000)發現,EPCs在胎盤囊的三層結構中均呈現高豐度。其中,EPCs-II(高表達C型凝集素)在外層富集,可能暗示其在早期聚集外部吸附物或抑制表皮細菌方面發揮作用。相比之下,EPCs-III(tfa+)和EPCs-IV(gata1+)在內層富集,與“鐵穩態”、“細胞遷移”及“血管發育”相關,其在雄性妊娠胎盤形成過程中可能參與侵襲和血管化過程。作為細胞外基質(ECM)的主要來源,FBs存在于中間層,富集“膠原蛋白生成”,可能促進胎盤囊結構形成和組織重塑。ICs則分散于三層結構中,調控免疫穩態。
基于scRNA-seq,scATAC-seq,空間轉錄組數據聯合分析,作者發現了具有干細胞潛能的”育兒袋上皮祖細胞(BEPCs)”。研究顯示,該類細胞在發育過程中與膠原蛋白基因呈現協同表達,并受到雄激素信號的強烈驅動。研究進一步證實,外源性雄激素處理可誘導雌性海馬形成育兒袋結構。由此,雄激素受體及其調控的育兒袋上皮祖細胞作為觸發育兒袋器官生成的關鍵起始因子。
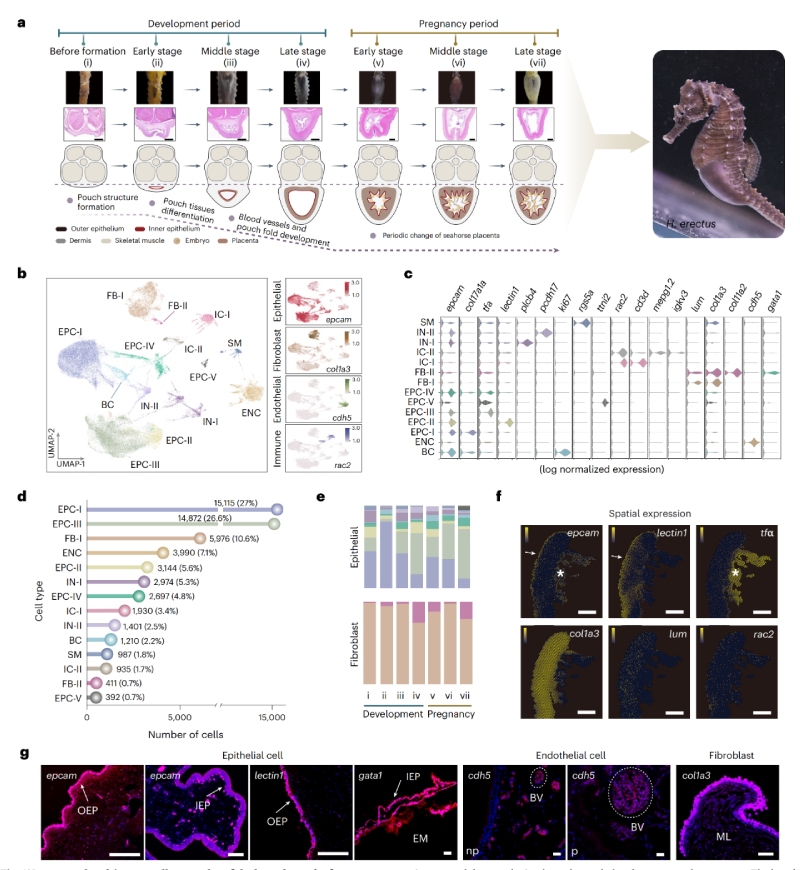
圖1?細胞圖譜構建
在雄性妊娠期間,海馬卵囊內層會發生顯著的形態學改變并呈現肥大,形成類似胎盤的組織結構,在細胞譜系轉變過程中,多個海馬特異性基因(如pastn1、sp-chia)在偽胎盤形成中起關鍵作用。作者發現EPC-I(muc15)、EPC-I(cxcl14)和EPC-IV在人類中與絨毛外滋養層細胞(EVTs)及絨毛滋養層細胞(VCTs)具有高度相似的基因表達譜,海馬雄性妊娠和哺乳動物常規雌性妊娠的胎盤形成過程,可能具有趨同的細胞機制和調控基礎。海馬缺乏foxp3基因(哺乳動物中調控Treg細胞的關鍵基因),但可能通過其它免疫細胞(如cd4+il2rb+?Tregs?和巨噬細胞)維持對胚胎的免疫耐受。
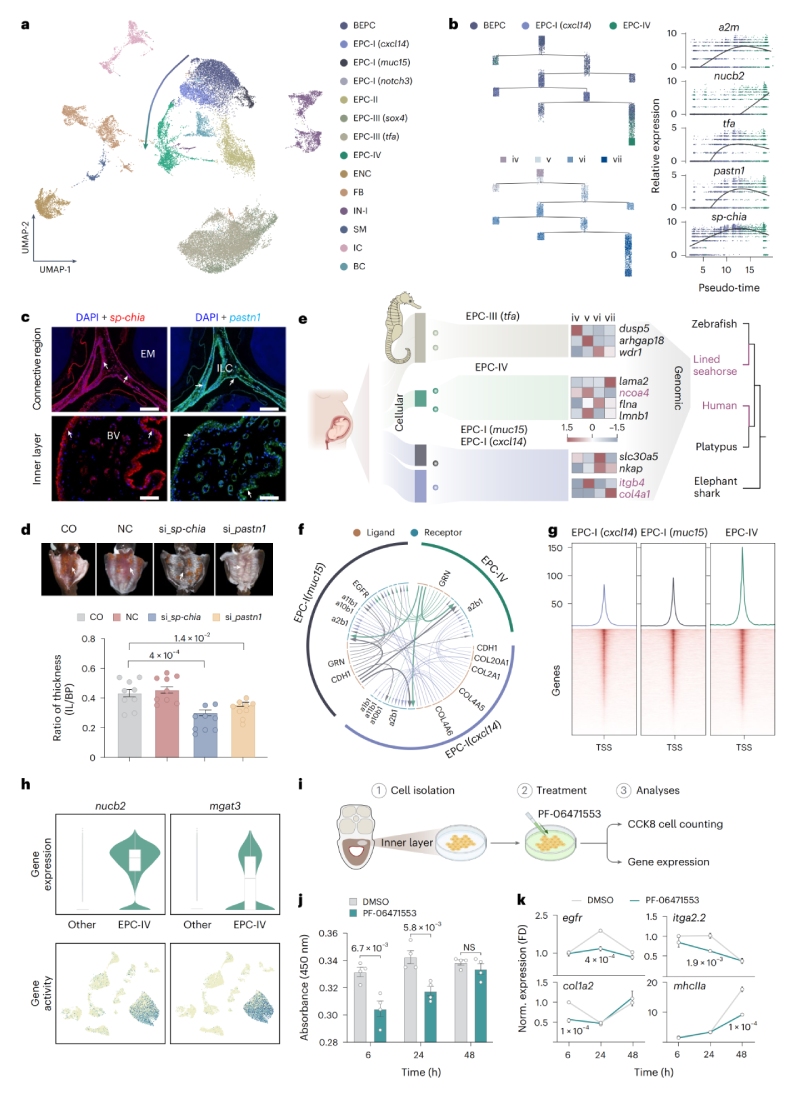
圖2?海馬假胎盤形成與子宮重塑的單細胞轉錄組分析圖譜
通過跨物種比較基因組學與單細胞多組學分析發現,盡管存在物種特異性差異,海馬假胎盤中的多數細胞類型在轉錄組特征上高度近似于人類的滋養層細胞——后者在調控胎兒生長及母體妊娠適應中發揮關鍵作用。其中,EPCs-II型細胞兼具表皮分化特征與凝集素介導的黏附功能,其可能是卵囊演化起源的重要線索。
進一步研究表明,海馬育兒袋與哺乳動物子宮在細胞和遺傳層面具有顯著同源性。海馬與哺乳動物生殖系統所呈現的趨同進化現象,可能源于特定細胞類型通過趨同演化形成相似的轉錄特征,進而實現類似功能,最終推動胎生機制的形成。
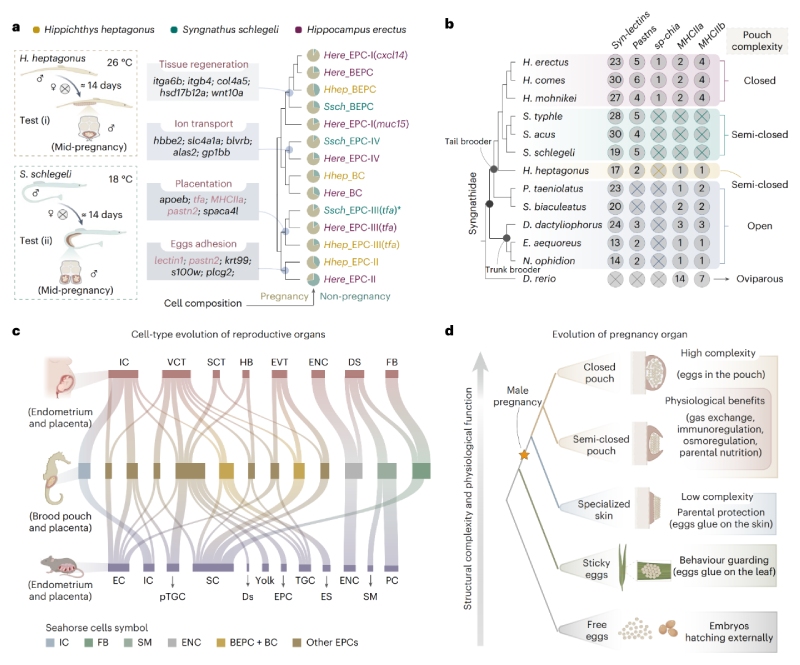
圖3?細胞類型進化分析
研究總結
該研究通過細胞分子與發育生物學的多維度分析,深入解析其卵囊發育與妊娠過程中的細胞遺傳動態。研究發現,卵囊形成的關鍵在于一種具有干細胞潛能的“育兒袋上皮祖細胞”群體。體內實驗證實,雄激素在卵囊形成中起主導作用。通過對其它動物的對比研究,作者揭示了海馬雄性親代撫育的早期進化機制,以及其細胞特征,存在與哺乳動物胎生動物相似的生命功能的趨同演化。
]]>研究背景
慢性肉芽腫病(Chronic?Granulomatous?Disease,?CGD)是一種由NADPH氧化酶2(NOX2)復合體功能缺失突變引起的原發性免疫缺陷病。這種缺陷會導致產生ROS的多種先天免疫細胞受到損害,機體抵御微生物的能力下降,導致CGD患者易發生反復感染和過度炎癥反應。本研究通過構建自然CGD小鼠模型,結合單細胞轉錄組(scRNA-seq)和空間轉錄組技術(包括BMKMANU?S1000平臺),深入解析了CGD肺部肉芽腫形成的細胞與分子機制,從CGD肺肉芽腫組織中鑒定出具有NOS2高表達特征的中性粒細胞和MMP12+巨噬細胞,并提出MIF和Morbidity為CGD治療的潛在靶點。
研究內容及結果
1、通過接觸清潔環境(CL)建立CGD肉芽腫模型
通過不同環境暴露(SPF或CL)實驗,系統研究了環境病原體對CGD遺傳背景小鼠表型的影響。表現為HE染色全肺葉顯微圖像可見CL環境下的Ncf2?/?小鼠肺部出現明顯的肉芽腫結構,伴隨炎癥細胞浸潤和組織壞死,其他組小鼠肺部正常。免疫熒光顯示肉芽腫中的巨噬細胞(CD68+)表達上皮標志物E-cadherin,提示巨噬細胞發生上皮樣轉化。通過ELISA法檢測CL?Ncf2?/?小鼠血清中的IL-1β和IFN-γ炎癥相關蛋白水平較CL?WT組增加。16S?rRNA/ITS?測序顯示CL?Ncf2?/?小鼠肺部細菌(如克雷伯菌和葡萄球菌)和真菌(如Talaromyces)負荷顯著增加。
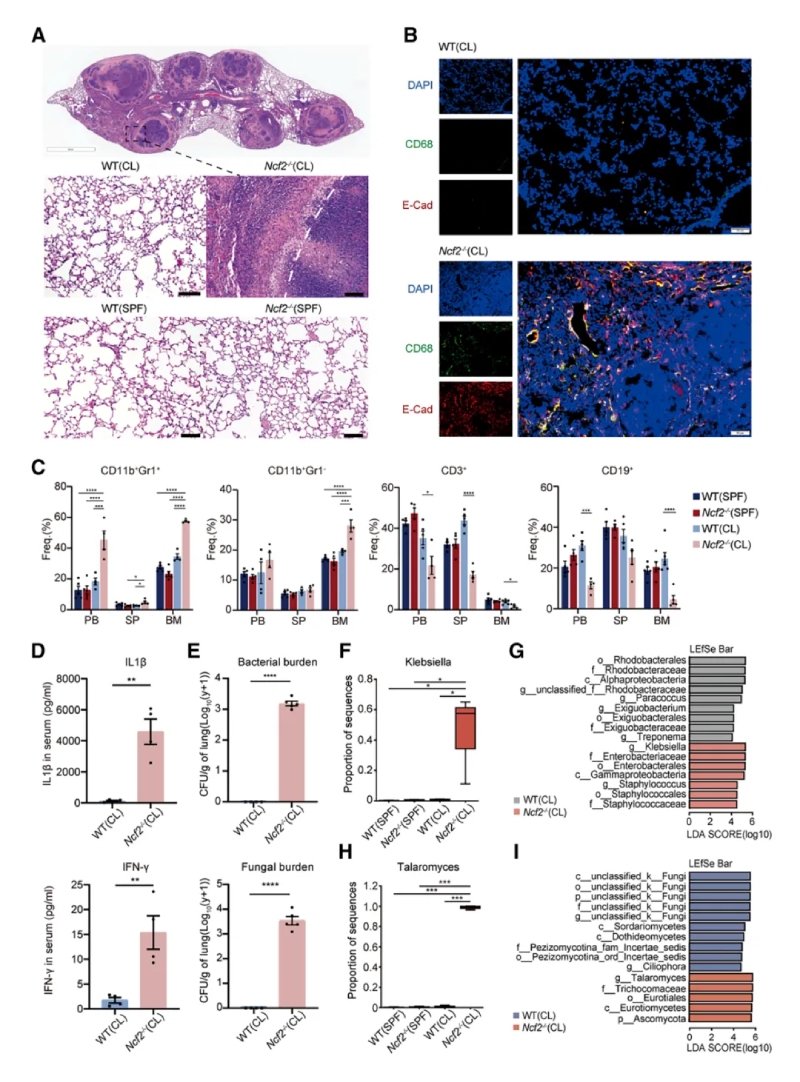
2、單細胞分辨率下小鼠肺部免疫景觀的分析
通過單細胞RNA測序和流式細胞術分析發現CGD小鼠肺部炎癥微環境以髓系細胞浸潤為主,提示其參與肉芽腫形成。其中單細胞測序,最終保留了25296個高質量細胞樣本,每個細胞平均包含6466條比對讀數和1972個基因,并識別出9種免疫細胞類型及九種基質細胞類型。還發現一個以細胞周期相關基因(Mki67和Top2a)高表達為特征的細胞簇。通過流式細胞術分析發現,CL?Ncf2?/?小鼠中CD11b高表達的F4/80+MDMs在比例和絕對數量上均顯著增加,這些結果揭示了慢性肉芽腫性葡萄膜炎(CGD)小鼠在肺部感染過程中免疫細胞組成的顯著變化,尤其是中性粒細胞和MDMs的明顯聚集現象。
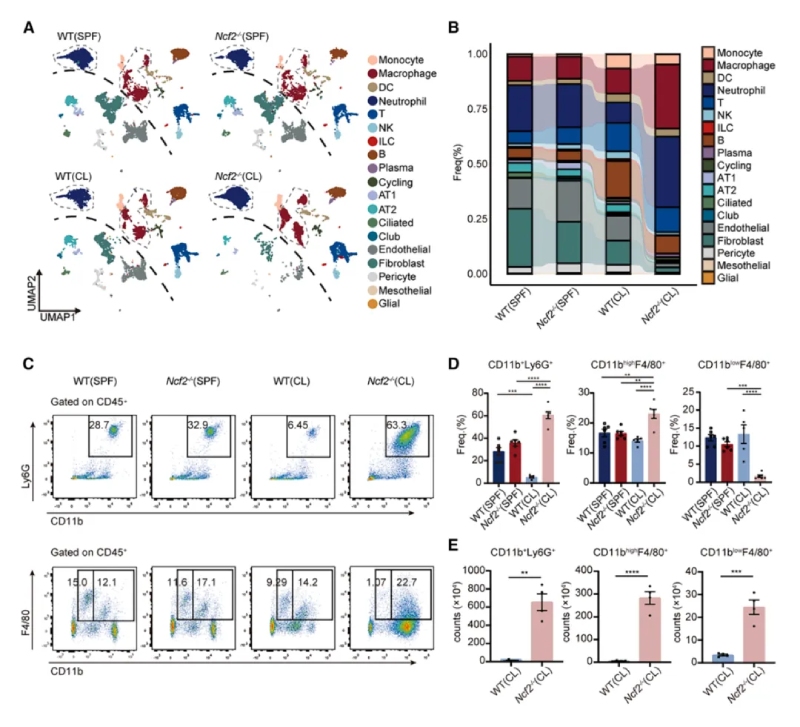 3、CGD小鼠中性粒細胞的轉錄改變
3、CGD小鼠中性粒細胞的轉錄改變
研究顯示,中性粒細胞分為6個亞群,其中Neu6亞群在CL?Ncf2?/?小鼠中顯著擴增且Neu6高表達促炎基因(如Nos2、Mif、Tnf、Il1b)。NO是一種在病原體清除過程中起關鍵作用由NOS2合成的可擴散小分子,CL?Ncf2?/?小鼠肺部的總NO生成量顯著升高。流式驗證顯示CL?Ncf2?/?小鼠肺中NOS2high中性粒細胞比例顯著增加。這些結果均提示NOS2high中性粒細胞通過產生一氧化氮(NO)和促炎因子,驅動肉芽腫核心區的炎癥反應。
 4、CGD小鼠單核細胞-巨噬細胞區室的轉錄改變
4、CGD小鼠單核細胞-巨噬細胞區室的轉錄改變
通過對巨噬細胞亞群進行聚類分析和功能富集,發現巨噬細胞分為5個亞群,Mac1亞群在CL?Ncf2?/?小鼠中顯著增加。Mac1同時表達M1(Nos2)和M2(Arg1)標志物,以及促纖維化基因(如Mmp12、Spp1)。免疫熒光顯示MMP12+巨噬細胞特異性分布于肉芽腫外圍。以上結果提示MMP12+巨噬細胞具有混合表型,可能通過細胞外基質重塑促進肉芽腫的纖維化包裹。
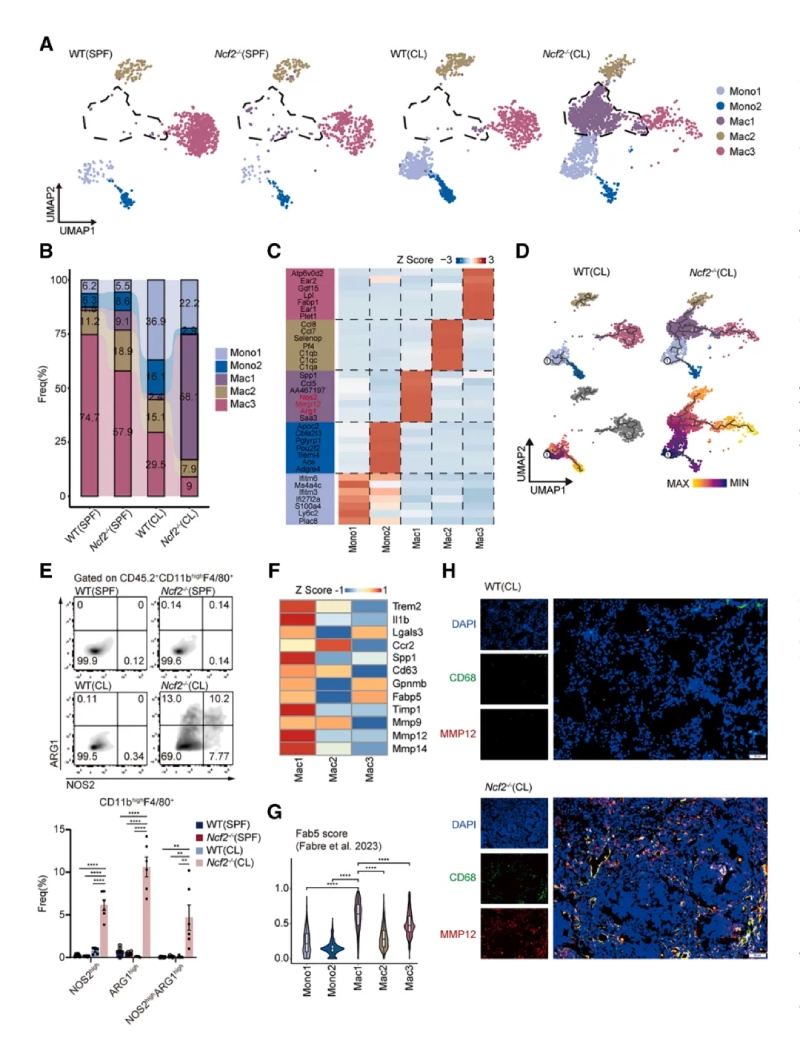 5、空間轉錄組揭示肉芽腫結構
5、空間轉錄組揭示肉芽腫結構
使用SeekSpace和BMKMANU?S1000平臺分析肺組織切片的空間轉錄組。研究表明肉芽腫核心區富集中性粒細胞(Neu6亞群),外圍富集巨噬細胞(Mac1亞群)和成纖維細胞。Mac1高表達促炎(Nos2)和促纖維化(Mmp12、Fn1)基因。成纖維細胞高表達膠原基因(Col1a1、Col4a1),提示其參與纖維化。

以上結果提示通過BMKMANU?S1000平臺進行的空間轉錄組發現Neu6占據肉芽腫中心區域,而Mac1和成纖維細胞在外圍區域參與細胞外基質重塑和纖維化包被過程。明確了肉芽腫中不同細胞的分布和功能分工,揭示了炎癥與纖維化的協同作用。
6、干預策略的效果驗證
通過對巨噬細胞亞群進行聚類分析和功能富集,發現巨噬細胞分為5個亞群,Mac1亞群在CL?Ncf2?/?小鼠中顯著增加。Mac1同時表達M1(Nos2)和M2(Arg1)標志物,以及促纖維化基因(如Mmp12、Spp1)。免疫熒光顯示MMP12+巨噬細胞特異性分布于肉芽腫外圍。以上結果提示MMP12+巨噬細胞具有混合表型,可能通過細胞外基質重塑促進肉芽腫的纖維化包裹。
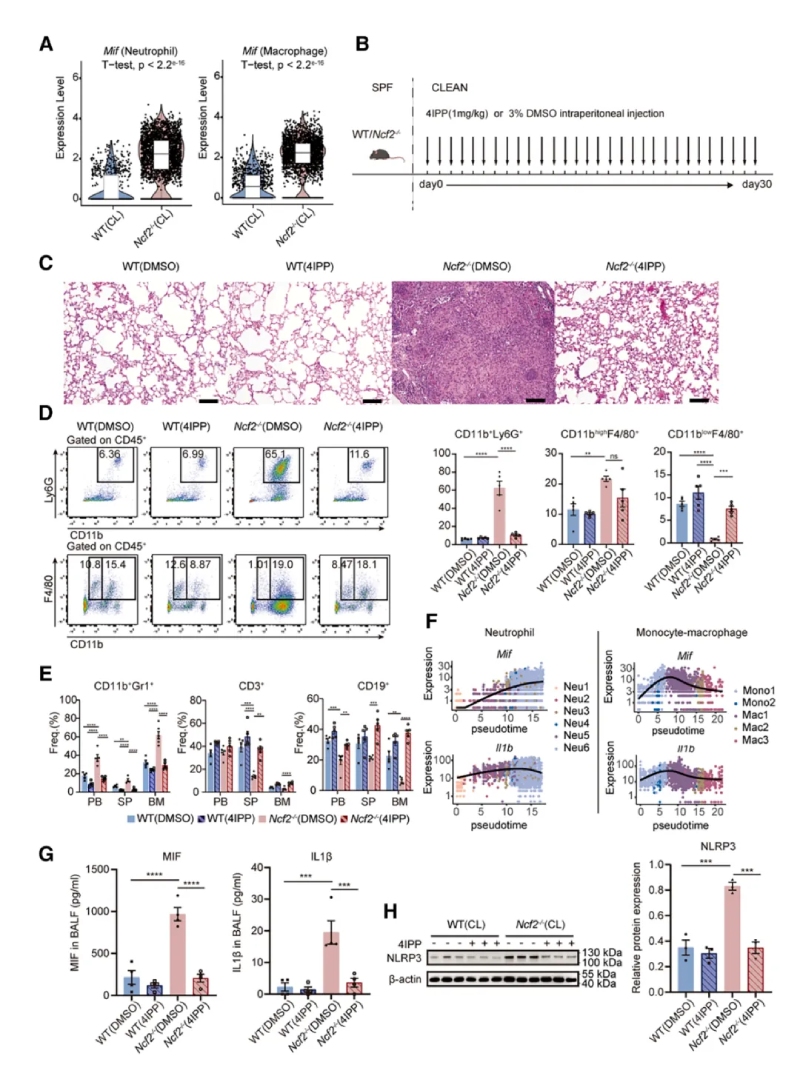
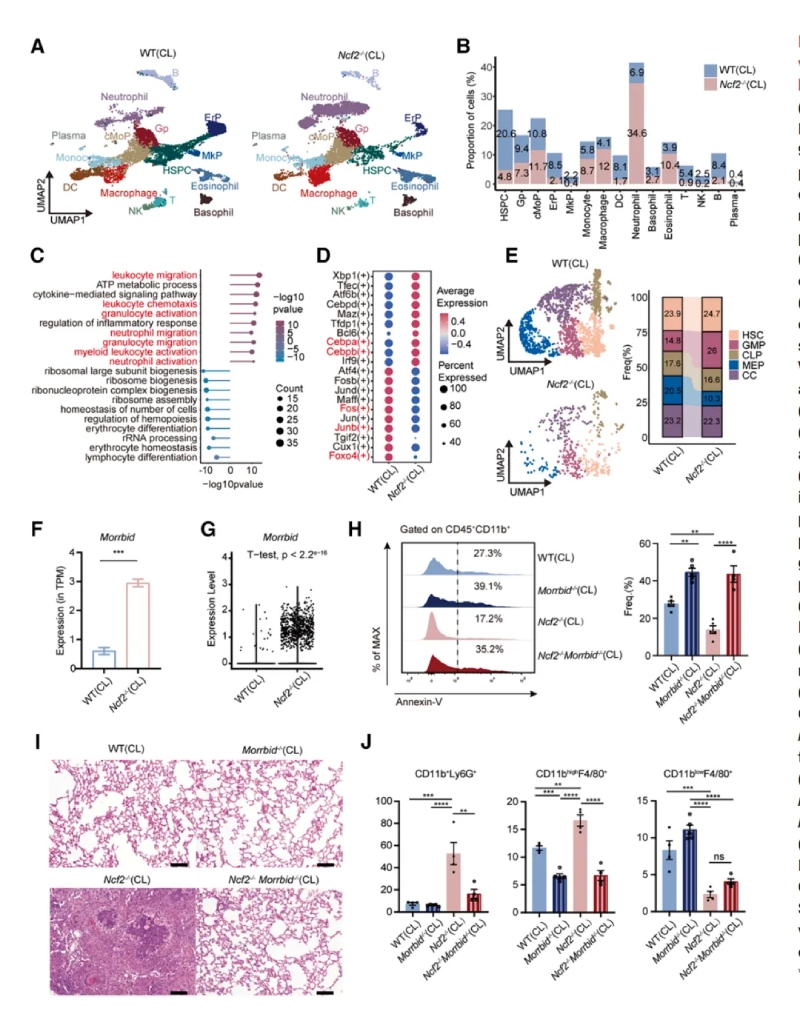 研究總結
研究總結
首次通過環境暴露構建自然CGD模型,結合多組學技術解析肉芽腫的細胞和分子機制。本研究揭示了CGD肉芽腫中NOS2high中性粒細胞和MMP12+巨噬細胞的協同作用,并提出MIF和Morrbid作為潛在治療靶點,為CGD的精準干預提供了理論基礎。BMKMANU?S1000空間轉錄組通過高分辨率空間定位明確揭示了中性粒細胞(Neu6亞群)在肉芽腫核心、巨噬細胞(Mac1亞群)和成纖維細胞在外圍的分布。在基因表達驗證時確認了NOS2、MIF、IL-1β等基因在肉芽腫不同區域的差異表達,這一結果與scRNA-seq結果高度一致,展示了BMKMANU?S1000在疾病微環境研究中的高效性。
]]>環境配置
1. conda安裝
wget?https://repo.anaconda.com/miniconda/Miniconda3-py39_4.12.0-Linux-x86_64.sh
下載完成之后運行(按提示安裝)
sh Miniconda3-py39_4.12.0-Linux-x86_64.sh
安裝完成后執行以下命令
source ~/.bashrc
2. 環境配置
conda create -n (環境名) python=3.9
激活環境:
conda activate (環境名)
添加鏡像源
conda config –add channels?https://mirrors.bfsu.edu.cn/anaconda/cloud/bioconda/
conda config –add channels?https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge/
conda config –add channels?https://mirrors.bfsu.edu.cn/anaconda/pkgs/free/
conda config –add channels?https://mirrors.bfsu.edu.cn/anaconda/pkgs/main/
查看鏡像源
conda config –show channels
3. 安裝python模塊
conda install numpy=1.23.4
conda install opencv=4.7.0
conda install pandas=2.2.2
conda install matplotlib=3.8.4
conda install tqdm=4.66.4
conda install seaborn=0.12.2
conda install tifffile
conda install scanpy=1.9.8
4. 軟件安裝
conda install samtools=1.18 sambamba=1.0.1 bedtools=2.31.1
conda install macs2=2.2.7.1
conda install deeptools=3.5.5
conda install ucsc-bedgraphtobigwig=472
conda install bwa=0.7.17
conda install bioconda::cutadapt=5.0
conda install bioconda::ucsc-bedsort
conda install bioconda::ucsc-bedclip
5. 安裝指定R版本(v4.3)
conda install -c conda-forge r-base=4.3.1
6. 安裝指定R包
conda install -c conda-forge r-signac=1.14.0
conda install -c conda-forge r-seurat=4.3.0
conda install bioconda::bioconductor-rtracklayer=1.62.0
conda install -c conda-forge r-kableextra=1.4.0
conda install -c conda-forge r-optparse=1.7.5
conda install -c conda-forge r-this.path=2.7.0
conda install conda-forge::r-r.utils
conda install conda-forge::r-devtools=2.4.5
options(timeout = 9999999999999)
devtools::install_github(“davidsjoberg/ggsankey”)
install.packages(‘DT’)
參考基因組配置
參考基因組需要使用bwa進行建庫(流程使用bwa比對)
gtf文件中需包含gene_id,gene_name,gene_biotype/gene_type
人和小鼠基因組推薦使用:
https://www.10xgenomics.com/support/software/cell-ranger-arc/latest/release-notes/reference-release-notes
refdata-cellranger-arc-GRCh38-2020-A-2.0.0
refdata-cellranger-arc-mm10-2020-A-2.0.0
其他基因組可自行選擇版本。
配置文件填寫
配置文件可參考config.txt進行填寫,內容說明如下:
## fq數據路徑,FQ1為read1文件路徑,FQ2為read2文件路徑
FQ1 /path/to/read_1.fq.gz
FQ2 /path/to/read_2.fq.gz
## 芯片解碼文件路徑
FLU /path/to/flu_info.txt
## 組織明場圖片路徑
HE /path/to/HE.tif
#INSIDE 1 #是否扣除組織內部空白區域,默認1扣除,選0為保留
## 組織熒光圖片路徑
#FL /path/to/FL.tif #組織熒光圖,和HE圖二選一
#FLC -1 #熒光圖顏色通道,值為0,1或2,選擇-1為自動判斷
## 組織區域文件路徑
JSON /path/to/roi.json #如不給定,則流程會自動使用上面的圖片進行識別,自動識別建議使用熒光圖
## 參考基因組文件
GenomeVer xxxx #版本信息,出現在報告中
Ref /path/to/ref/genome.fa #基因組fa文件
Gtf /path/to/ref/gene.gtf #基因組gtf文件
SPECIES human #物種文件,人和小鼠分別為human/mouse,其他物種自行給定
## 輸出結果及輸出文件前綴
OUTDIR /path/to/result/dir/
PREFIX outfile-prefix
## barcode 類型,S3000芯片無特殊情況為V2
BCType V2
## 線程數
Threads 8
## cs序列類型,無特殊情況為cs12
CSType cs12
軟件運行
在配置好的環境中,運行命令:
/path/to/BSATAC -c config.txt -s 0
其中 -c 是配置文件,-s 是所選擇運行的步驟,0是全部運行
結果說明
outdir/
├── 01.fastq2BcUmi # barcode識別目錄
├── 02.LinkBcChip # 芯片barcode對應
├── 03.AllheStat # 多級分辨率和組織區域處理
├── 04.Cutadapt # 接頭過濾
├── 05.Mapping # reads比對結果
├── 06.Fragment # Fragment分析
├── 07.WebReport # 網頁版報告
├── BSTViewer_project # 收集的結果目錄
└── prefix # 芯片原始barcode結果
聯合分析操作指南
注意說明,當前分析需要百創智造的空間RNA和空間ATAC分析流程輸出的結果!!!
空間RNA和空間ATAC的主要結果各自存儲在BSTViewer_project文件下。
1. FiJi對齊
1.1:百創智造空間ATAC與空間轉錄組聯合分析之切片對齊概述
https://zhuanlan.zhihu.com/p/1903040885328945604
1.2:百創智造空間ATAC與空間轉錄組聯合分析之切片對齊前圖像生成-01
https://zhuanlan.zhihu.com/p/1898370805919903880
1.3:百創智造空間ATAC與空間轉錄組聯合分析之FIJI中手動對齊切片-02
https://zhuanlan.zhihu.com/p/1903079101796941928
1.4:百創智造空間ATAC與空間轉錄組聯合分析之切片上點的映射-03
https://zhuanlan.zhihu.com/p/1903081341429452952
1.5:百創智造空間ATAC與空間轉錄組聯合分析之Level1向其他level水平的點映射-04
https://zhuanlan.zhihu.com/p/1907854262597321846
其它: Fiji的下載與安裝(windows 10)
https://zhuanlan.zhihu.com/p/18027378723
特殊說明:軟文詳細講解了對齊的原理,腳本實現以及結果說明。其中,軟文中涉及的腳本存在?[SpatialATAC/v1.0.2/fragment/ImageJ_FJ] 中。
2. 聯合分析
2.1 配置文件說明
配置文件可參考config.yaml?[SpatialATAC/v*/fragment/Joint_analysis/v*/]?進行填寫,內容說明如下:
ATAC和RNA分析路徑(一級目錄下)
ATAC_analysis path/BSTViewer_project/
RNA_analysis path/BSTViewer_project/
######映射文件(通過Image Fiji獲取)(默認讀取L18 L9 L7 L5四個水平的結果)Fiji path/
輸出路徑 outdir path/analysis
##其它參數 alpha 0.1 ##透明度,范圍是0-1 logfc 0.1
2.2 流程執行
perl Joint_main_v1.2.pl -c config.yaml -s 0
其中 -a -b -c 是配置文件,-s 是所選擇運行的步驟,0是全部運行
perl Joint_main_v1.2.pl
Usage:
-a spatial config ##空間轉錄組主流程的配置文件
-b atac config ##空間ATAC主流程的配置文件
-c joint config ##聯合分析的配置文件(見2.1)
-s run step
step number:
0: run all step
1: Joint analysis
2: Joint report
3: total report
2.3 結果說明
L*_alignment/
├── L*_Joint_marker_gene.csv ##聯合分析marker基因
├── L*_joint.rds ##rds文件
├── L*_Sankey_plots.pdf ##桑基圖
├── L*_Sankey_plots.png
├── L*_heatmap.pdf ##marker基因熱圖
├── L*_heatmap.png
├── L*_CoveragePlot.pdf ##基因組瀏覽器圖
├── L*_CoveragePlot.png
├── L*_Spatial_ATAC.cluster.pdf ##ATAC聚類圖
├── L*_Spatial_ATAC.cluster.png
├── L*_Spatial_Joint.cluster.pdf ##聯合分析聚類圖
├── L*_Spatial_Joint.cluster.png
├── L*_Spatial_RNA.cluster.pdf ##RNA聚類圖
└── L*_Spatial_RNA.cluster.png
**特殊說明**
1. L*_joint.rds 中存在3個assay,分別是:ATAC(peak矩陣),Spatial(表達矩陣),RNA(活性矩陣)。
2. 結果生成2個報告:聯合分析的報告,以及Spatial、ATAC以及聯合分析的匯總報告
中文題目:5-ASA的腸道微生物代謝降低了其在炎癥性腸病中的臨床療效
發表期刊:nature medicine
發表時間:2023
影響因子:82.9
研究背景
炎性腸病 (IBD)是一種慢性、使人虛弱的胃腸疾病,治療失敗率較高。目前尚無系統的方法預測對IBD療法的反應。抗炎藥物美沙拉嗪,也被稱為5-氨基水楊酸 (5-ASA),是IBD最常用的處方療法之一,通常在結腸內發揮作用;然而,隨著時間的推移,超過一半的IBD患者對5-ASA沒有反應或最終失去反應。因此,有必要確定并消除此類治療失敗的原因。
研究思路
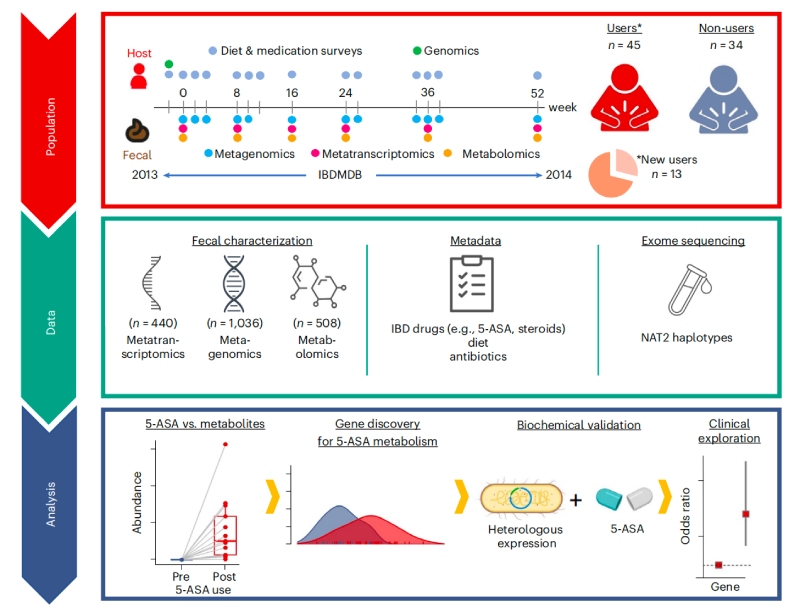
研究重點
1、來自IBD患者的多組學研究確定了5-ASA的使用
利用人類微生物組項目炎癥性腸病多組學數據庫(IBDMDB,http://ibdmdb.org),對79名克羅恩病(CD)或潰瘍性結腸炎(UC)患者的糞便樣本進行多組學分析,共鑒定了1036個宏基因組(MGX)、440個元轉錄本(MTX)和508個非靶向代謝組(MBX),以及283個MBX-MGX對和213個MGX-MTX對。隨后針對13名新使用5-ASA的患者來確定5-ASA在體內直接調節的特定糞便代謝物,結果發現5-ASA使用前后中的5-ASA和N-乙酰5-ASA水平呈顯著差異,且經5-ASA治療后,賴氨酸合成途徑中的細菌代謝產物—2-氨基己二酸的含量減少,煙酸代謝也發生了顯著變化。
進一步評估微生物、宿主和其他因素對這些差異顯著的代謝物的相對貢獻,最終發現5-ASA的藥物水平在確定5-ASA調節代謝物水平方面具有最大的預測能力(35%),其次是微生物組特征(15%)和其他宿主因素(7%)。隨后,通過另一個獨立的IBD患者隊列中鑒定并驗證了兩種可能的5-ASA衍生物—N-丙酰基5-ASA和N-丁酰基5-ASA,它們的變化可以部分地由腸道微生物組來解釋。

圖1?5-ASA可直接影響糞便代謝組,并通過微生物組進行生物轉化
2、5-ASA代謝腸道微生物酶的鑒定
接下來確定參與5-ASA生物轉化的腸道微生物酶。首先對服用及未服用5-ASA的患者微生物進行轉錄組分析,最終鑒定了兩個顯著過表達的具有推定乙酰轉移酶功能的UniRef90基因簇:GNAT家族NAT(UniRef90 ID: C7H1G6)和乙酰輔酶a乙酰轉移酶(UniRef90 ID: R6TIX3),以及四個具有乙酰輔酶a乙酰轉移酶結構域的候選基因(IDs: R6CZ24, R5CY66, T5S060和A0A1C6JPG6)。隨后將糞便樣本分為N-乙酰5-ASA高水平和N-乙酰5-ASA低/陰性水平兩組,計算與N-乙酰5-ASA相關的每個元轉錄組基因簇的敏感性和特異性,結果發現了另外7個假定的乙酰轉移酶基因簇與使用者的N-乙酰5-ASA水平呈正相關。最終得到的12個之前未被表征的候選乙酰轉移酶可分為硫解酶和酰基輔酶A N-酰基轉移酶兩組,其中酰基輔酶A NAT家族比硫解酶家族表現出更大的序列異質性,且酰基輔酶A NAT酶幾乎都來自擬桿菌門,而硫解酶來自厚壁菌門。通過研究菌株水平的基因組,同樣發現了一部分普氏鐮孢菌株編碼相關的乙酰轉移酶。
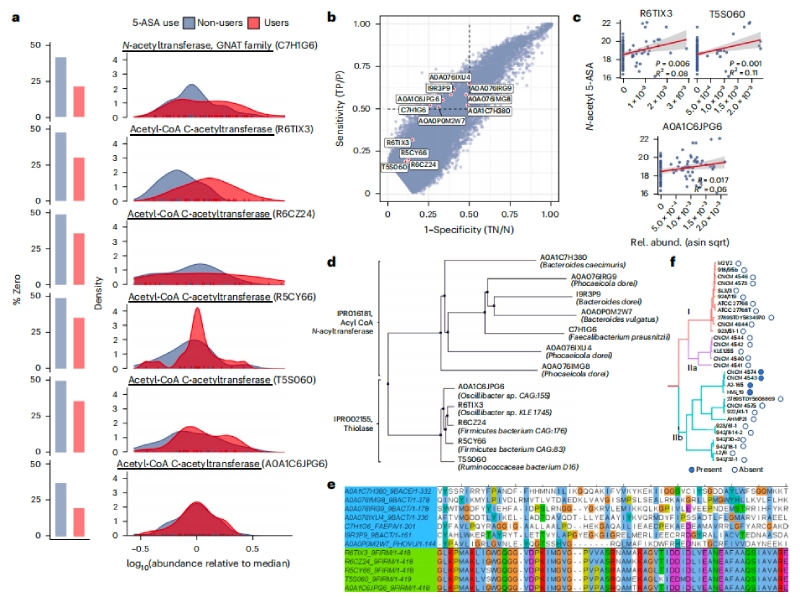
圖2N-乙酰5-ASA微生物酶包括硫代酶和酰基輔酶A N-乙酰轉移酶
3、5-ASA失活酶生化特性的推定
為了推測硫解酶和酰基輔酶A NAT超家族具有潛在的5-ASA滅活能力,選用來自厚壁菌門的候選硫解酶CAG:176(UniRef90 ID: R6CZ24)和來自福氏假單胞菌的酰基輔酶A NAT(UniRef90 ID:C7H1G6)用于進一步的生化表征。用已知的腸道鏈球菌酶作為陽性對照,對乙酰化5-ASA采用體外質譜分析,最終證實了厚壁菌CAG:176硫解酶和F. prausnitzii酰基輔酶A NAT具有使用乙酰輔酶A乙酰化5-ASA的能力。
隨后測試了硫解酶對其他含胺基異生素(包括5-ASA異構體4-ASA、異煙肼、普魯卡因胺和肼屈嗪)的乙酰化活性,結果并未發現這些化合物的任何乙酰化,這表明了硫解酶的底物選擇性。此外,編碼第二個預測的硫解酶基因(R6TIX3)的Oscillibacter sp菌株KLE 1745的活培養物也能夠將5-ASA乙酰化為N-乙酰基5-ASA。
為了深入了解厚壁菌CAG:176硫解酶(FcTHL)如何乙酰化5-ASA,將FcTHL的乙酰化未配體晶體結構疊加在乙酰化硫解酶晶體結構上,與來自充分表征的革蘭氏陰性分枝菌(ZrTHL)的乙酰輔酶A復合。隨后將FcTHL與NAT (PDB ID: 2PFR)的晶體結構進行比對,進一步探究厚壁菌CAG:176硫解酶的推定活性位點是否類似于腸道鏈球菌NAT的活性位點,最終揭示了活性位點區域中的半胱氨酸和組氨酸三聯體。
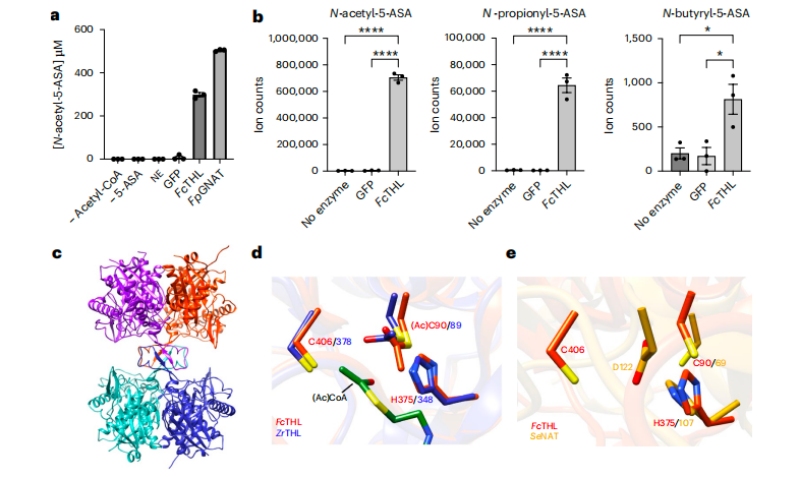
圖3?5-ASA在體外的乙酰化活性的證實
4、IBD治療失敗與5-ASA代謝酶的宏基因組攜帶有關
基于臨床服用5-ASA的個體差異性,接下來檢查12種乙酰轉移酶的存在是否與5-ASA治療失敗有關。收集39名在任何時間點接受5-ASA治療及使用類固醇患者的609份糞便樣本,使用多變量邏輯回歸模型,調整與疾病風險相關了因素-年齡、性別、吸煙狀況和IBD亞型,納入每個參與者的NAT2表型來解釋宿主遺傳,最終發現糞便樣本中存在4種乙酰轉移酶基因與類固醇起始風險增加顯著相關。4種乙酰轉移酶基因有三種來自硫解酶超家族,一種來自酰基輔酶A超家族。且較早的發病年齡和CD與UC狀態相比,同樣與開始使用類固醇的更高風險顯著相關。但未發現類固醇使用風險和大腸桿菌NAT的宏基因組存在聯系。
在敏感性分析中,微生物乙酰轉移酶基因數量的增加與類固醇起始風險的增加顯著相關;從未使用過5-ASA治療的患者基因家族與類固醇的使用沒有正相關。隨后使用混合效應模型對參與者進行調整,發現存在三個或更多乙酰轉移酶基因也與類固醇使用風險增加相關,這在隨后在未使用類固醇的208名5-ASA使用者隊列中得到了驗證。

圖4腸道微生物5-ASA滅活乙酰轉移酶與5-ASA使用者治療失敗的更大風險相關
研究總結
本研究結果首次提供了特異性腸道代謝酶與IBD 5-ASA治療失敗之間的直接聯系,從而產生了直接的臨床潛力。5-ASA是UC最常用的處方藥。為了保持有效,它必須以未修飾的形式存在于結腸腔中,因此,與其他藥物不同,它在小腸中的吸收很差。5-ASA治療失敗的個體通常進展為風險更高的免疫抑制治療,而只有在必要時 (即腸道微生物組中存在修飾5-ASA的硫解酶序列)才有能力這樣做,這將為精準醫學提供有價值的生物標志物。此外,闡明微生物失活5-ASA的酶學機制可能有助于未來研發微生物特異性酶抑制劑,以增強5-ASA的療效。在本研究中,我們的數據是觀察性的;需要更多的介入研究來加強我們的發現,無論是通過在IBD患者中的臨床試驗還是通過單定植小鼠。目前,這些發現為IBD的個性化微生物醫學打開了一扇概念之窗。
]]>研究單位:西安交通大學基礎醫學院院長張保軍教授團隊
期刊名:cellular & molecular immunology
影響因子:21.8
樣本類型:8-12周齡小鼠
文章采用技術:空間轉錄組測序、單細胞RNA測序(scRNA-seq)
引言
在對抗感染和腫瘤的免疫戰場上,記憶?CD8+?T?細胞是守護機體的「長效衛士」,?它們能快速識別再次入侵的病原體或癌細胞,發動強力攻擊。長久以來,樹突狀細胞(DC)被認為是主導記憶?T?細胞分化的核心「指揮官」。研究人員通過整合單細胞RNA測序(scRNA-seq)和空間轉錄(BMKMANU?S1000)技術,揭示了單核細胞才是調控記憶?CD8+?T?細胞分化的關鍵「操盤手」,其奧秘藏在「細胞接觸」與「分子信號」的雙重機制中。其中BMKMANU?S1000空間轉錄組學平臺大放異彩,成為揭CCR2+單核細胞促進記憶CD8+?T細胞分化的關鍵工具。
研究背景
CD8+?T細胞在適應性免疫中扮演著關鍵角色,能夠保護機體免受病原體感染和消除惡性細胞。當CD8+?T細胞識別到抗原后,會迅速增殖并分化為效應CD8+?T細胞和記憶CD8+?T細胞。效應CD8+?T細胞負責即時清除感染,而記憶CD8+?T細胞則提供長期保護,能夠在再次遇到相同抗原時迅速啟動免疫反應。然而,單核細胞(monocytes)作為重要的髓系免疫細胞,雖在炎癥遷移中作用明確,但其是否直接參與T細胞分化尚無定論。
研究策略
動物模型與樣本制備
實驗動物:優先選擇8-12周齡的CD45.1+/CD45.2+及OT-I轉基因小鼠
樣本制備:脾臟單細胞懸液通過機械分離和紅細胞裂解(ACK緩沖液)制備
技術手段
- 流式細胞術分析
細胞分選:從感染LM-WT的小鼠脾臟中分選出cDCs、moDCs和單核細胞等抗原呈遞細胞(APCs)
細胞刺激與培養:將分選出的APCs與OVA肽段體外共孵育后,過繼轉移到已接受初始OT-I+?CD8+?T細胞的WT受體小鼠體內,或與體外刺激的初始CD8+?T細胞共培養
表型檢測:通過流式細胞術檢測CD8+?T細胞的分化表型,包括記憶相關標記物(如cKit、Sca1、Bcl6、TCF1和Eomes)的表達
- 空間分辨轉錄組學
樣本處理:對感染第0天和第5天的小鼠脾臟組織進行切片,并進行H&E染色以確定組織形態學特征
空間轉錄組學分析:利用BMKMANU?S1000空間轉錄組學技術對脾臟切片進行分析,揭示不同免疫細胞亞群在脾臟中的空間分布
數據整合:將scRNA-seq數據與空間轉錄組學數據進行整合,通過Seurat等工具的FindTransferAnchors和TransferData函數,將scRNA-seq中識別的細胞類型映射到空間轉錄組學數據中,實現細胞類型的空間定位
- 免疫熒光染色
樣本處理:對感染第0天和第5天的小鼠脾臟組織進行切片,并進行免疫熒光染色
抗體選擇:使用特異性抗體標記CD8+?T細胞、單核細胞(CCR2+)、樹突狀細胞(CD11c+)等關鍵細胞類型
結果分析:通過顯微鏡觀察并拍攝染色切片,分析不同細胞類型在脾臟中的共定位關系
- 生物信息學分析
差異基因分析:利用Wilcoxon秩和檢驗等統計方法,識別不同細胞亞群間的差異表達基因
通路富集分析:通過ClusterProfiler等工具對差異表達基因進行通路富集分析,揭示潛在的生物學過程
偽時間分析:利用Monocle3等工具構建細胞分化軌跡的偽時間軸,分析CD8+?T細胞亞群在分化過程中的基因表達變化
研究結果
研究結果一:通過空間分辨轉錄組學繪制急性感染后脾組織的圖譜
為了研究免疫反應過程中不同免疫細胞的空間分布和CD8+?T細胞分化的調節機制,研究人員通過整合單細胞RNA測序(scRNA-seq)和BMKMANU?S1000空間轉錄組(ST)技術,分析了急性感染模型中脾臟免疫細胞的空間分布與CD8??T細胞分化機制。實驗采用OT-I轉基因小鼠模型,通過李斯特菌(LM-OVA)感染誘導免疫反應,關鍵發現包括:1)脾臟形成7個功能集群(B細胞、T細胞、單核/巨噬細胞等);2)感染后T/B細胞區擴大,APCs(B細胞、DC、巨噬細胞)在T細胞區外圍聚集;3)空間重組與T細胞分化(MP/TE亞群)顯著相關。多組學整合揭示了免疫微環境的動態調控機制,為理解感染中細胞互作提供了多組學視角。
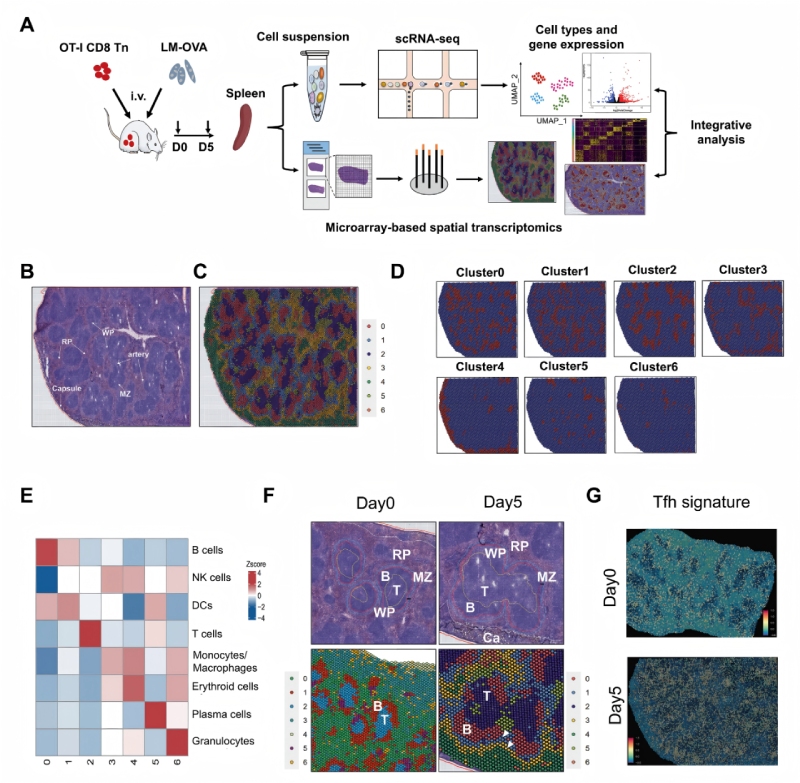
研究結果二:急性感染期間效應和memoryCD8?T細胞分化軌跡不同
為了探索CD8+?T細胞分化的空間決定因素,特別是與近端細胞的相互作用,對于急性感染模型研究人員進行了亞群分析、分化路徑分析和功能驗證,結果表明在急性感染中,IFN反應型CD8+?T細胞和CD8+MP?細胞之間分化軌跡的不同意味著,效應和記憶CD8+?T細胞的命運決定于免疫反應的初始階段,而IFN應答和MP?CD8+?T細胞分別是效應和記憶CD8+?T細胞分化途徑的初始階段。
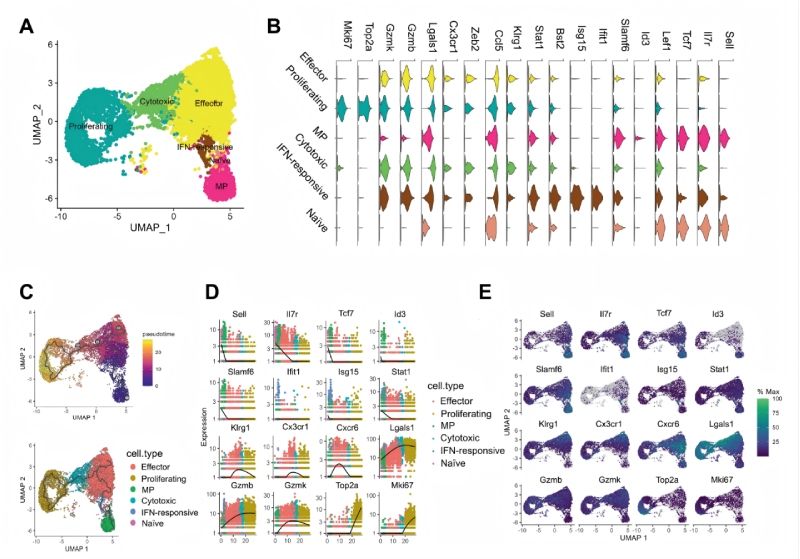
研究結果三:跨組織區域細胞亞群的鑒定和空間圖譜
為了研究脾臟免疫細胞在感染前后的動態變化,研究人員通過整合單細胞RNA測序(scRNA-seq)和BMKMANU?S1000空間轉錄組(ST)技術,進一步鑒定了供體和受體脾臟中的免疫細胞群。結果顯示:①細胞組成:從1.6萬細胞中鑒定出19類,包括CD8??T細胞亞群(如記憶前體和IFN反應性細胞)及髓系細胞。②動態變化:感染后第5天,T細胞區從以初始CD8+?T細胞為主轉為記憶前體和效應細胞主導,且單核細胞取代DC成為主要浸潤的髓系細胞。③功能提示:不同的APC和CD8+?T細胞之間的相互作用在免疫反應和CD8+?T細胞的分化中起著重要作用。
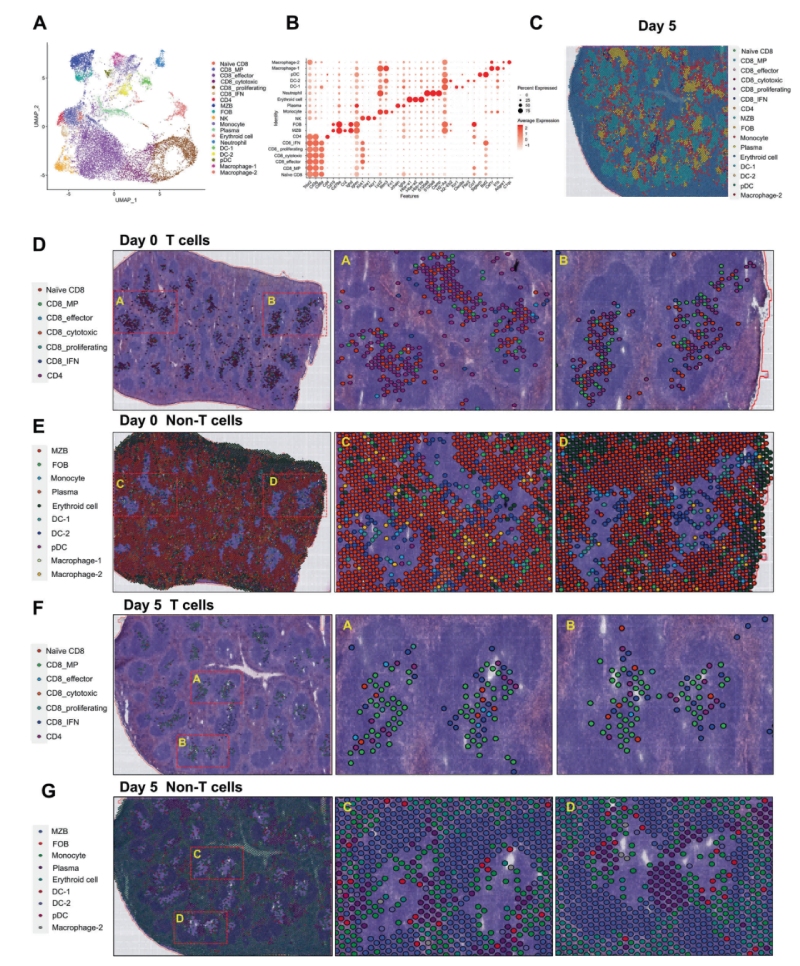
研究結果四:單核細胞和CD8+?MP細胞的抗原依賴性共定位
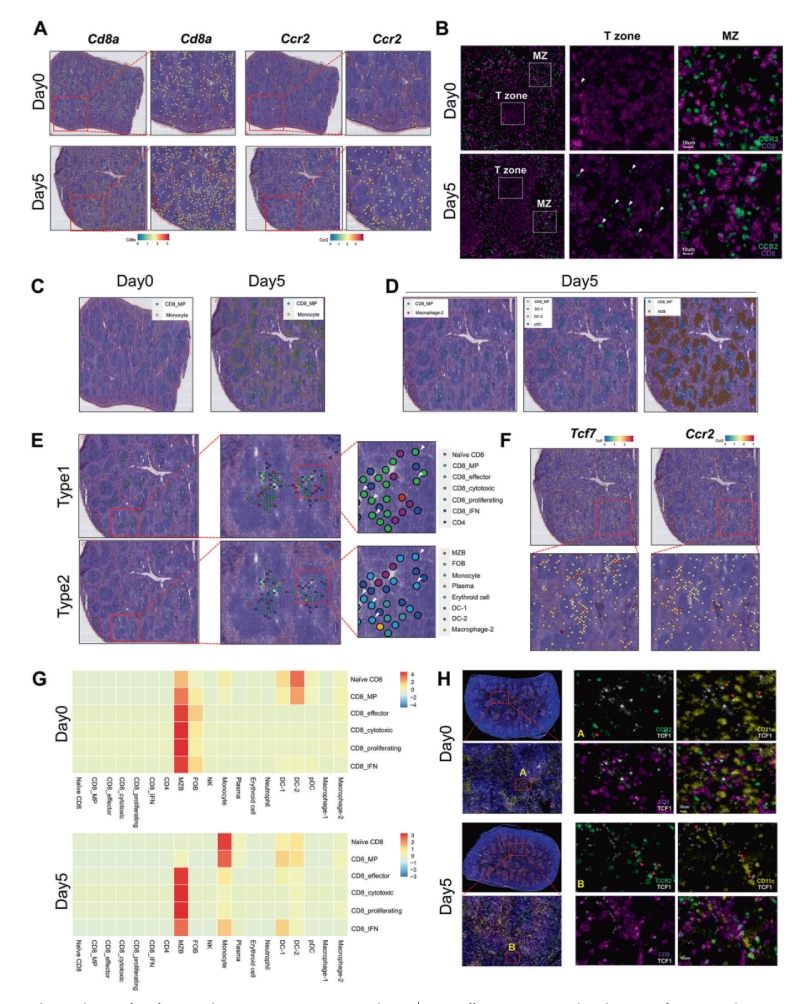
為了探索不同細胞類型之間潛在的細胞相互作用,研究人員通過評估特異性免疫細胞標記物的表達、免疫熒光染色及空間轉錄組分析,檢查了第0天和第5天脾臟中CD8+?T細胞和單核細胞的空間分布。結果顯示單核細胞和CD8+?MP細胞以抗原刺激依賴性的方式在空間上共定位,表明單核細胞可能對CD8+?MP細胞的分化有關鍵影響。
研究總結
CD8+T?細胞是適應性免疫的重要執行者;特別是記憶CD8+?T細胞對強效和長期保護至關重要。CD8+?T細胞分化在轉錄水平上的調控機制已被廣泛研究。然而,人們對感染過程中效應和記憶CD8+?T細胞分化的空間要求仍然知之甚少。研究人員通過整合BMKMANU?S1000空間轉錄組(ST)技術和scRNA-seq技術,發現了記憶CD8+?T細胞分化的新機制,該機制涉及單核細胞和CD8+?MP細胞之間的解剖學鄰近性和TGF-β信號傳導。該研究結果闡述了記憶CD8+?T細胞命運決定的新機制,并強調單核細胞作為關鍵的APC群體,通過細胞間接觸依賴的方式在感染過程中促進記憶CD8+?T細胞的分化。
]]>
先睹為快:新一代植物廣靶定量代謝技術
本次發布的植物廣靶定量代謝組學產品并非簡單技術迭代,百譜生物研發經理李天運在發布會中詳細介紹了該產品是針對植物研究領域長期存在的檢測靈敏度不足、數據庫不全、缺少內標定量三大痛點,形成多維度解決方案,獲與會專家高度認可。

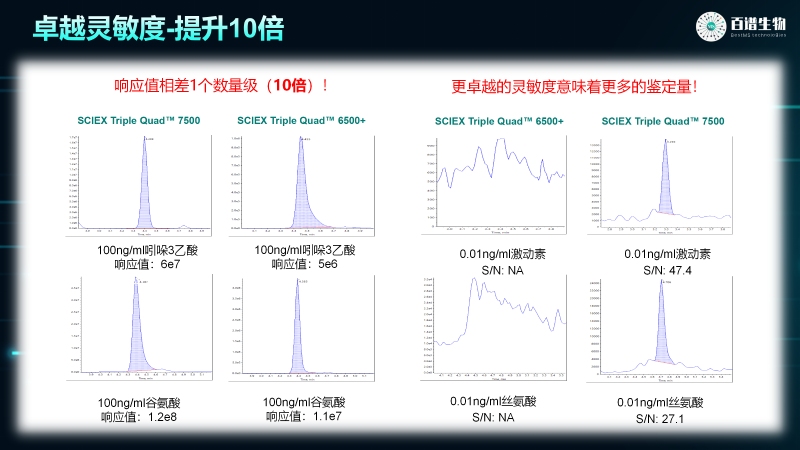
高靈敏檢測:突破微量代謝物局限
針對傳統技術難以捕捉植物抗逆、品質形成過程中微量代謝物的痛點,該產品采用 AB SCIEX QTRAP7500 高靈敏度檢測平臺,將檢測限降低一個數量級,可精準定量低豐度植物代謝物、特殊滲透調節物質等低含量成分。即使是植物應對脅迫時微量積累的關鍵代謝物,也能被清晰捕捉。
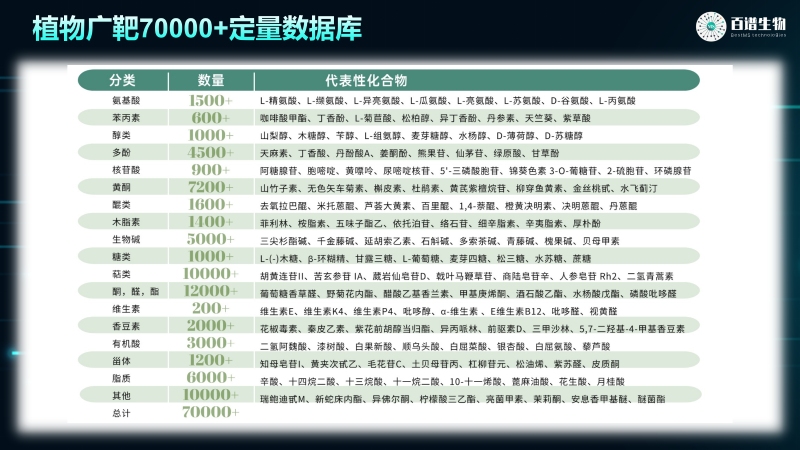
數據庫完整性:高達7萬種代謝物
代謝組研究的核心在于數據庫完整性,該產品將代謝物數據庫從傳統 “數千種” 擴容至 “數萬種”,高達7萬種,并補充標準品多維注釋信息,使代謝物鑒定準確率大幅提升,可有效避免 “漏檢關鍵成分”“誤判物質種類” 等問題。
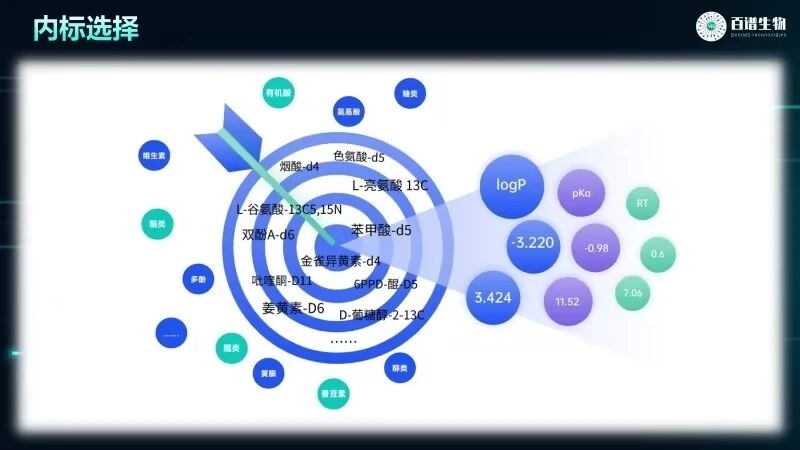
業內首發:同位素內標定量
業內首次在植物廣靶技術上加入同位素內標進行定量分析平臺,擁有多種內標選擇,能根據不同的檢測需求和化合物特性,靈活挑選合適的內標,通過基質內標線性和自研算法校正,可以得到更為準確的定量結果。
SCIEX中國市場開發經理方晶晶深度解讀了QTRAP7500設備技術的革新和優勢,他認為:“代謝物的高通量、高靈敏和大隊列穩定性是未來代謝組發展的重要方向”。

企業戰略布局:推動代謝組技術從實驗室走向田間
發布會上,百邁客生物創始人兼CEO鄭洪坤指出行業核心痛點:“測序能找植物的基因,但代謝組才懂它的‘品質’!”他表示,現在代謝組在農業里的用處還沒完全打開,未來要靠 “測序 + 代謝組” 組合拳,比如在育種環節通過基因篩選鎖定潛力品種,再以代謝組驗證維生素含量等品質指標,提升育種效率與準確性。

百譜生物總經理曹以襯公布近期發展規劃:這款植物廣靶定量代謝組學產品,是我們團隊潛心研發的核心成果——它打破了傳統技術在檢測靈敏度、數據庫覆蓋度上的局限,將成為植物研究領域的高效工具!未來我們將聯合頂尖設備廠商升級檢測平臺,進一步提升超微量代謝物檢測能力,為農業科研創新、品種改良與產業升級提供關鍵支撐。

專家實踐驗證:技術落地解決產業實際難題
中國科學院海洋研究所段德麟研究員表示:傳統品質研究像“看表面癥狀”,難知代謝層面連鎖反應;定量代謝組技術能幫我們“找病根”:可定量脅迫下小分子代謝物變化,整合數據構建完整代謝調控網絡,還能測低含量抗逆物質提供精準標志物,培育口感更好、風味更佳的品種。
北京大學現代農業研究院王旭教授以辣椒研究為例:“傳統依賴感官評價判斷辣椒辛辣度,精準度低且主觀性強。該技術可定量檢測辣椒素合成的多種中間產物,我們已通過其篩選出維生素C高含量的辣椒品種,推動品質育種升級。”
中國科學院海洋研究所張立濤副研究員認為:傳統方法研究微藻蝦青素合成途徑,僅能發現終產物變化,無法定位影響關鍵代謝物的中間途徑,尤其是強光、氮源等因素調控不同產物的分支途徑。植物廣靶定量代謝組學技術可精準識別終產物及其中間產物,為提升微藻中活性物質合成提供了明確的方向。

北京大學現代農業研究院王旭教授(左二)、中國科學院海洋研究所段德麟研究員(右二)、中國科學院海洋研究所張立濤副研究員(右一)
新聞相關報道

?9?月?17?日,百邁客生物推出的植物廣靶定量代謝技術,標志著植物代謝組學正式進入廣靶、精準、實用的新階段,未來將在功能食品開發、作物育種優化、農業抗逆管理等多個方面發揮深遠影響。>>詳情

2025?年?9?月?17?日,由百邁客生物與百譜生物聯合主辦的?“植物廣靶定量代謝組學發布會”?在青島成功舉辦。會上推出的植物廣靶定量代謝組學技術,為植物基礎研究與農業產業升級注入新動能,引發行業廣泛關注。中國科學院海洋研究所段德麟研究員、北京大學現代農業研究院王旭教授等專家出席。>>詳情

2025 年 9 月 17 日,在青島舉辦的“植物廣靶定量代謝組學發布會”上。百邁客生物與百譜生物聯合推出植物廣靶定量代謝組學技術,為植物基礎研究與農業產業升級注入新動能,引發行業廣泛關注。中國科學院海洋研究所段德麟研究員、北京大學現代農業研究院王旭教授等專家與企業代表應邀出席。>>詳情

9月17日,新一代植物廣靶定量代謝組學技術在青島自貿片區發布,該技術使得植物在抗逆、品質形成過程中微量積累的關鍵代謝物得以清晰捕捉,為深入研究植物生理機制注入新的科研動力。中國科學院海洋研究所段德麟研究員等行業專家受邀出席。>>詳情

9月17日,百邁客生物在青島自貿片區發布新一代植物廣靶定量代謝組學技術,該技術直面植物研究領域長期存在的三大核心難題:檢測靈敏度低、代謝物覆蓋范圍有限、缺乏精準定量的內標方法,通過多維創新提升植物代謝組學研究的效率與準確性。>>詳情

2025年9月17日,百邁客生物與百譜生物聯合發布“植物廣靶定量代謝組學技術” 。該技術采用?AB SCIEX QTRAP7500?高靈敏度檢測平臺,將檢測限降低一個數量級,相較于傳統?SCIEX Triple Quad? 6500 +?平臺,對低豐度植物代謝物、特殊滲透調節物質等低含量成分的檢測能力顯著提升。為深入研究植物生理機制提供更多可能。>>詳情

9月17日,百邁客生物與百譜生物聯合發布“植物廣靶定量代謝組學技術” 。這一技術使得植物在抗逆、品質形成過程中微量積累的關鍵代謝物得以清晰捕捉,為深入研究植物生理機制提供了技術可能。中國科學院海洋研究所段德麟研究員、北京大學現代農業研究院王旭教授等行業專家及企業代表受邀出席。>>詳情

植物研究領域受限于檢測靈敏度不足、代謝物數據庫不全、缺乏精準內標定量等問題,尤其是在非模式植物研究中,微量代謝物難以捕捉、關鍵成分易漏檢誤判、定量結果準確性不足等痛點,嚴重制約了研究效率與產業應用進程。此次發布的植物廣靶定量代謝組學技術,針對性提出多維度解決方案。>>詳情
]]>
植物代謝組研究伴隨著分析技術的發展和對植物代謝認識的深入而逐步開展。19世紀末,科學家開始研究植物的化學成分及其藥理作用,這成為植物代謝組研究的雛形。
1998年,Steven Oliver 在一篇關于酵母功能基因組學的綜述文章中創造了 “metabolomics” 一詞。此后,世紀之交發表的多篇論文闡述了代謝物譜分析在植物代謝調控等研究中的應用潛力,這些研究主要采用非靶向分析方法,推動了多元統計方法在植物和細胞功能研究中的廣泛應用。
2010年前后,植物代謝組學研究日趨成熟,高分辨質譜技術、代謝組學成像技術和代謝組學定量分析技術等得到廣泛應用。
2023年,中國科學院分子植物科學卓越創新中心李軒團隊構建了涵蓋苔蘚植物、石松植物、蕨類植物、裸子植物和被子植物五大類群的150多個植物物種的參考代謝組及其數據庫RefMetaPlant,為植物代謝及植物生理相關研究提供了關鍵支持,促進了代謝組學領域的數據交流共享與合作。
量溯植物代謝·解碼植物生命
從種子萌發到果實成熟,植物每一步代謝活動都暗藏“量化密碼”。植物代謝組是植物生長發育、逆境響應及品質形成的?“動態檔案”,而定量分析則是解讀這份檔案的關鍵鑰匙。為此,由青島百譜生物科技有限公司、北京百邁客生物科技有限公司聯合主辦的“植物廣靶定量代謝組學發布會”,將于9月17日在山東青島隆重召開。
發布會以“量溯代謝” 為核心,將系統揭示植物定量代謝組學在科學研究與農業產業中的雙重驅動力。發布會將分享基于液相色譜-高分辨質譜(LC-HRMS)等技術的植物代謝物定量方法創新,詳解如何實現對數千種初生代謝物(如糖、氨基酸)和次生代謝物(如黃酮、生物堿)的精準定量與動態追蹤。
此外,本次發布會將匯聚來自全國的諸多杰出學者與專家,他們將以論壇交流的形式在此深入研討與溝通,共同促進代謝組學在農業中的研究,為我國在該領域的科技進步貢獻卓越的智慧和力量。
出席嘉賓
鄭洪坤:百邁客生物創始人兼CEO
曹以襯:青島百譜生物科技有限公司總經理
段德麟:中國科學院海洋研究所研究員
王 ?旭:北京大學現代農業研究院研究員
張立濤:中國科學院海洋研究所副研究員
方晶晶:SCIEX中國,生命科學研究市場開發經理
李天運:青島百譜生物科技有限公司質譜研發經理
?會議流程
時間? ? ? ? ? ? ? ? 會議環節
14:00~14:30 ?實驗室參觀
14:30~14:35 ?歡迎詞
14:35~15:05 ?從“廣篩”到“精量”:植物廣靶定量代謝組技術–解鎖植物代謝研究全新維度
15:05~15:35 ?SCIEX定量質譜驅動高覆蓋靶向代謝組學研究
15:35~15:45 ?產品發布儀式
15:45~16:30 ?主題討論會
16:30~17:00 ?新品煥新&互動贏好禮
新品煥新·互動贏好禮
為方便更多同仁參與,屆時將開啟線上同步直播。本次發布會特別為線上同仁打造 “專屬福利環節”,讓您在云端參會也能收獲滿滿、驚喜不斷!
- 將帶來植物代謝組學核心產品深度拆解—— 不僅詳細講解產品功能與技術優勢,更會結合科研場景、產業需求,精準匹配您的實操難點與痛點,幫您快速判斷產品如何助力研究與生產;
- “互動贏好禮” 重磅福利同步上線!只需在直播間實時參與交流:無論是針對技術/產品提出疑問、圍繞話題分享觀點留言,還是搶答專家拋出的關鍵問題,只要積極融入答疑討論,就能獲得抽獎資格,贏取多重豐厚精美大獎。讓您在吸收行業前沿干貨的同時,輕松抱走驚喜好禮,線上參會也能 “滿載而歸”!
歡迎各界人士關注“百邁客生物”視頻號,積極報名預約共赴這場植物代謝組學的學術盛宴!
]]>

本次調研由北京科技創新促進中心黨委書記李萍帶隊,科技服務業部部長付文均、城市科技部部長曲宏,以及北京醫藥健康科技發展中心前沿部部長李潛等領導共同參與;百邁客生物聯合創始人王瑞、財務負責人蔣躍征等陪同接待,并與調研團隊進行座談交流。

座談會上,王瑞首先對市科委領導一行的到來表示熱烈歡迎,并圍繞百邁客生物的發展歷程、核心業務板塊、核心技術優勢,以及未來在基因科技領域的長期戰略布局作詳細介紹。她強調,百邁客生物始終以?“技術創新驅動產業發展”?為核心理念,深耕生命科學領域,致力于為生命科學領域提供高質量的產品與服務。
隨后,市科委各位領導結合各自分管領域,針對百邁客生物的發展痛點與未來方向,提出了兼具專業性與指導性的建議:
李萍書記對百邁客生物在基因科技領域的科技創新突破及產業轉化方面取得的成果給予高度肯定。她明確表示,市科委將進一步加大對科技服務型企業的扶持力度,尤其在研發投入、人才引進、平臺建設等方面提供更多政策賦能,助力企業攻克技術難關、擴大發展規模。
李潛部長聚焦醫藥健康領域前沿技術的政策導向,結合行業人才需求趨勢,為百邁客生物解讀了最新政策支持方向,幫助企業更好把握行業機遇、應對發展挑戰。
付文均部長圍繞?“科技服務業與企業需求深度融合”?的核心,提出了具體建議。
曲宏部長則結合百邁客生物的技術特點,從科技應用場景拓展、產學研合作等方面,提供了系統性的指導思路,為企業拓寬發展邊界提供參考。
此次調研不僅深化了市科委對百邁客生物發展現狀與核心需求的精準了解,更為企業后續在政策對接、資源整合、戰略規劃等方面提供了明確的方向和支持等方面提供了明確的方向指引與支持。百邁客生物將以此為契機,進一步加強與政府部門的常態化溝通協作,加速科技創新與成果轉化,為推動北京國際科技創新中心建設、助力基因科技行業高質量發展積極貢獻企業力量。
]]>發表時間:2024年12月
合作單位:農業農村部農業環境保護研究所
發表期刊:Journal of Hazardous Materials(IF 8.9)
研究背景
塑料污染現狀嚴峻且向納米級轉化:塑料已成為現代生活不可或缺的材料,但大量塑料廢棄物造成了全球緊迫的環境問題,在水、海洋生物、人類排泄物甚至極地沉積物等多種介質中均有檢出。環境風化作用使塑料碎片逐漸分解為微塑料(<5μm)和納米塑料(<1μm),其中納米塑料(NPLs)因更豐富、反應性更強,能到達更偏遠區域并穿透活細胞,環境風險遠高于微塑料。納米塑料與抗生素抗性基因(ARGs)的關聯研究不足:已有研究表明塑料可促進 ARGs 增殖,且納米材料(如金屬納米顆粒 AgNPs、ZnO NPs)與細菌抗生素抗性進化相關,但關于納米塑料對細菌抗生素抗性的影響尚不明確。金屬納米顆粒與納米塑料在物理化學性質和界面活性上存在本質差異,且雖有研究指出微塑料可吸附 ARGs 加速其傳播,但納米塑料與耐藥菌共存狀態及作用機制的研究仍較零散。
粘質沙雷氏菌的研究價值:粘質沙雷氏菌在自然環境中分布廣泛,可定殖于水和土壤介質,是醫院獲得性感染的重要條件致病菌,能引發肺炎、尿路感染和敗血癥等疾病,且其自身攜帶 ARGs。隨著廣譜抗生素的大量使用,該菌的耐藥性增強,對臨床治療構成挑戰,因此研究納米塑料脅迫下其抗生素抗性軌跡具有重要意義。
測序策略
轉錄組測序(RNA-seq)+基因組測序
研究結論
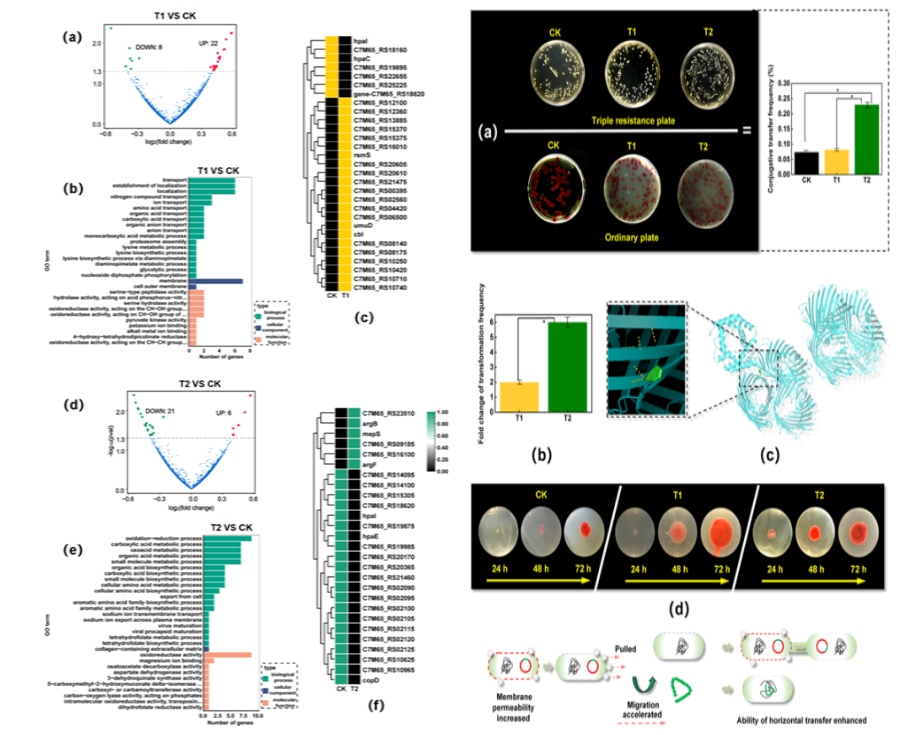
1、納米塑料可促進粘質沙雷氏菌抗生素抗性進化,且作用受粒徑影響:
暴露于納米塑料后,粘質沙雷氏菌對磺胺甲噁唑(SMX)、諾氟沙星(NOR)、鏈霉素(STR)、卡那霉素(KA)等抗生素的抑菌圈逐漸縮小,ARGs 相對豐度顯著高于無納米塑料組(CK),且呈現 “200nm 納米塑料組(T2)>600nm 納米塑料組(T1)>CK” 的規律(T2、T1、CK 的 ARGs 相對豐度中位數分別為 0.82%、0.62%、0.56%)。
隨著傳代,CK 和 T1 組的 ARGs 豐度呈下降趨勢(歸因于基因適應性成本),但 T1 組下降幅度小于 CK 組,而 T2 組 ARGs 豐度呈上升趨勢,表明小粒徑納米塑料對細菌抗生素抗性進化的促進作用更顯著。
2、不同粒徑納米塑料促進抗性進化的機制存在差異:
600nm 納米塑料:主要通過影響細菌細胞膜通透性促進抗性。轉錄組分析顯示,其誘導的差異表達基因(DEGs)多與物質運輸和細胞膜功能相關,如 LysE 家族轉運蛋白、ABC 轉運蛋白通透酶等表達上調,增強抗生素外排能力,進而提升細菌抗性。
200nm 納米塑料:主要通過調控細菌代謝過程增強抗性。其誘導的 DEGs 多參與氧化還原過程、羧酸代謝過程等,如硫胺素焦磷酸結合蛋白(TPP)下調導致細菌代謝受損,激活抗性相關基因表達;環氧奎奴基還原酶 QueH 上調提高細菌蛋白質合成效率,促進外排泵等抗性相關蛋白合成。同時,200nm 納米塑料對細菌生長抑制更強,使細菌進入低代謝休眠狀態,抵抗抗生素攻擊。
3、納米塑料通過誘導基因突變增強細菌抗性:
納米塑料暴露會導致粘質沙雷氏菌發生單核苷酸多態性(SNP)突變,且粒徑越小突變概率越高(T2 組 SNP 純合子數量 31513 個,T1 組 31380 個)。突變基因功能主要涉及催化活性、結合、細胞膜、細胞過程和代謝過程,部分突變基因(如 HMI62_RS24365)可改變細胞膜功能,增強抗生素外排,進而提升抗性。
4、納米塑料可加速 ARGs 水平轉移,小粒徑作用更突出:
納米塑料暴露顯著提高粘質沙雷氏菌的接合轉移頻率(T2 組 22.98%、T1 組 8.13%、CK 組 7.35%)和游離質粒轉化能力(T2 組轉化頻率是 CK 組的 6 倍,T1 組是 CK 組的 2 倍),且轉移的質粒攜帶多種抗性基因和外排泵基因。
分子對接顯示,納米塑料可與細菌細胞膜蛋白(如 5NUQ,結合能 – 8.54kcal/mol)結合,像 “牽引器” 一樣促進細菌間接觸;同時,納米塑料能增強細菌運動能力(T2 組細菌擴散圈最大),進一步提高 ARGs 水平轉移頻率。
結果展示
現在,百邁客生物細菌基因組產品帶著五大核心升級重磅來襲,從樣本起始量到分析周期,從組裝方式到數據應用,全方位打破行業瓶頸,為你的科研加速!
升級 1:低建庫起始量,珍貴樣本不再 “難倒英雄漢”
??很多時候,科研中的細菌樣本來之不易 —— 可能是臨床分離的少量病原菌,也可能是環境中富集的稀缺菌株,樣本量不足往往讓后續實驗 “卡殼”。此次百邁客生物 產品針對性優化建庫技術:
- ONT 平臺:建庫起始量低至 500ng / 次,無需大量擴增即可啟動實驗;
- PB 平臺:建庫起始量低至 250ng / 次,珍貴樣本也能輕松滿足實驗需求。
再也不用為 “樣本少” 發愁,讓每一份寶貴樣本都能發揮最大研究價值!?
升級 2:純三代組裝,擺脫二代數據依賴,分析流程直接 “拎包即用”
傳統細菌基因組組裝常需依賴二代數據校正,不僅增加了實驗成本,還延長了分析周期,流程銜接中也容易出現問題。
百邁客生物此次實現僅三代組裝技術突破:無需搭配二代數據,僅憑三代長讀長優勢,就能完成高質量基因組組裝。更重要的是,我們已搭建好完備的純三代分析流程,從數據下機到組裝結果輸出,全程標準化、自動化,無需你額外調試,拿到樣本即可啟動分析,大幅減少流程搭建的時間成本。
升級 3:極速周期低至 15 天,科研進度 “快人一步”
科研競爭講究 “時間就是成果”,漫長的分析周期往往會錯過最佳研究窗口。
百邁客生物通過技術優化與流程重構,將細菌基因組項目的整體周期壓縮至低至 15 天—— 從樣本接收、建庫測序,到組裝分析、結果交付,全程高效推進,讓你更快拿到核心數據,加速論文撰寫與項目結題。
升級 4:成功率≥98%,超高成功率,實驗放心 “不翻車”
“樣本做廢了”“數據沒出來”,是科研中最讓人崩潰的情況。百邁客生物深知實驗可靠性的重要性,通過優化樣本提取、建庫緩沖體系等關鍵環節,將細菌基因組項目的整體成功率提升至≥95% ,顯著高于行業平均提取與實驗成功率。
無論是復雜細胞壁結構的細菌,還是低豐度樣本,我們都能最大限度保障實驗成功,讓你的科研每一步都更穩妥。
升級 5:個性化上云功能,數據挖掘 “更懂你”
誰說數據分析不能又快又聰明?百邁客生物獨家提供個性化云上服務,秒殺絕大多數同行!涵蓋六大功能,總有一款適合您:
百邁客生物打破這一局限,推出行業稀缺的個性化上云服務,五大核心功能直擊研究痛點,讓數據挖掘更高效、更精準。

百邁客生物會將您的項目報告推送到您的云賬號下,在項目下可以直接進行個性化分析,無需再整理復雜的文件表格進行輸入,無生物學基礎亦可輕松拿捏基因組個性化分析。
1、通用數據庫注釋篩選
基于云平臺,你可根據 “毒力基因”“耐藥基因”“代謝通路” 等研究關鍵詞,一鍵篩選目標注釋結果,無需在海量數據中手動查找;
打分參數、可靠性參數設置,均可隨性設置。
2、基因組圖譜查詢
云平臺內置【基因組圖譜展示工具】,點擊即可查看基因組環形圖、基因位置分布等信息,直觀掌握基因組結構(示意圖如下)

3、T3SS分泌系統預測
基于EffectiveT3精準預測III型分泌蛋白,致病機制研究更深入!
4、GTDB-tk分類鑒定
輸入細菌基因組序列,即可通過該工具完成物種分類,明確菌株進化地位
5、ANI分析
快速計算基因組間相似性,可選多參考基因組比對,并生成熱圖,物種界定更輕松!
6、SNP進化樹構建
物種進化樹(?稱系統發育樹或?命樹)是?物學中?來描述不同物種或?物類群之間演化關系的樹狀圖。它通過分?結構展示物種的分化過程、共同祖先及親緣關系的遠近。使?snippy軟件進?core snp 分析,基于樣本間的core snp ,使?phmyl構建進化樹,展示?標物種與給定的菌株之前的近緣關系。
用 snippy 篩選核心 SNP,結合 phmyl 構建進化樹,清晰展示目標菌株與參考菌株的親緣關系,助力溯源與進化分析。
從 “能做” 到 “做好”,從 “出數據” 到 “出成果”,百邁客生物細菌基因組產品始終以研究者需求為核心,不斷迭代升級。此次五大升級已全面落地,后續還將推出更多貼合科研場景的功能,為你的細菌研究提供更強大的技術支撐!
- 細菌基因組研究正不斷突破,百邁客生物還將持續推出更多升級服務!敬請期待!
- 無論是病原微生物研究、環境微生物組分析,還是功能基因挖掘,百邁客生物助您穩扎穩打、快人一步!
- 想了解更多產品細節,或預約樣本檢測,歡迎點擊下方按鈕,解鎖你的高效科研新方案~

]]>